This article discusses the embryologic development of the eye and orbital structures. Among the defects presented are anophthalmia and microphthalmia, coloboma, persistent hyperplastic primary vitreous, Coats disease, vascular malformations, encephalocele and nasolacrimal mucocele. Clinical and imaging features of the diseases are presented, along with radiographic images.
The eye forms directly from the brain early in the embryonic life by means of an outgrowth from the anterolateral portion of the embryonic neural tube. Malformations of the eye therefore may occur in isolation, accompany other complex malformations of the nervous system or may be a part of multisystem developmental abnormalities. Several genes linked with growth and development of the eye, as well as some specific embryologic defects have been identified. Developmental anomalies affecting vision, as well as those that cause visually obvious manifestations can be detected by clinical examination. However, many of the ocular and orbital anomalies require careful radiologic assessment to establish a definite diagnosis.
This article provides a brief discussion of pertinent embryology of the orbit as well as a review of the commonly encountered congenital malformations of the orbit.
Embryology
Development of the Ocular Globe
That the optic vesicle is derived from the forebrain was first established in 1817 by Pander. During the fourth week of development, on approximately day 22, the future eyes first appear as a pair of shallow linear grooves on either side of the developing forebrain. These optic sulci or optic grooves evaginating from the forebrain neural folds enlarge rapidly to form outpouchings called the optic vesicles and grow toward the surface ectoderm. As the expanded optic vesicle approximates the surface ectoderm (approximately on day 28), the distal surface of the optic vesicle called the retinal disc starts to invaginate, transforming into a goblet shaped optic cup.
Between day 31 and day 33 of development, the adjacent surface ectoderm starts to thicken to form the lens placode, which then invaginates and pinches off from the surface ectoderm during the fifth week to form the lens vesicle. The lens vesicle is a precursor to the solid lens of the eye. The double walled optic cup is connected to the forebrain vesicle by a narrow, hollow optic stalk. The invagination of the optic cup is see in the central portion and also a part of the inferior surface that forms a longitudinal groove called the choroidal fissure (also called the choroid fissure, the optic fissure and the retinal fissure). Eventually, the lips of the choroidal fissure fuse and the mouth of the optic cup becomes a round opening – the site of the future pupil.
The inner and the outer layers of the optic cup are initially separated by a lumen, the intra-retinal space, which will soon disappear with apposition of the 2 layers. The inner wall of the 2-layered optic cup develops into the neural retina, whereas/although the outer layer of the optic cup forms the melanin containing pigment layer of the retina. It has been suggested that a sheath of neural crest cells that envelop the optic vesicle receive contribution from the optic evagination and develop into the uveal pigment cells. The neural retina differentiates between the sixth week and the eighth month of fetal life. Axons from the neurons developing in the retina extend to the brain through the optic stalk, converting it to the optic nerve ( Fig. 1 ).
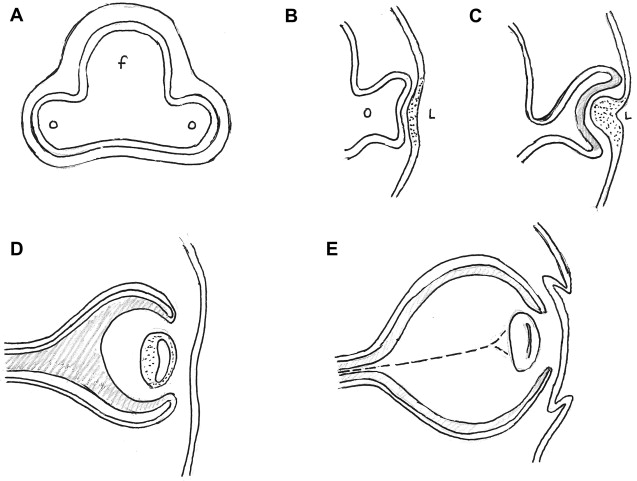
At the end of the fifth week, the primordial eye is surrounded by loose mesenchymal tissue that is derived in part from the neural crest. This sheath of mesenchymal tissue differentiates to form the inner choroid and outer sclera (comparable to the inner pia mater and outer dura mater of the brain). The sclera remains contiguous with the dura mater around the optic nerve. The pupillary muscles develop within this tissue from the underlying ectoderm of the optic cup. The pigment containing external layer of the optic cup forms the anterior surface of the iris. The anterior portion of the inner layer of the optic cup forms the inner layer of the iris and participates in the formation of the ciliary body as well. The pupillary muscles also form in this mesenchymal tissue. Anteriorly, the mesenchymal tissue splits into 2 layers, including a space that develops into the anterior chamber of the globe. The anterior layer of mesenchyme, along with the surface ectoderm forms the cornea, whereas/although the inner wall of the anterior chamber forms the pupillary membrane, which later disappears completely to form the pupil.
The mesenchymal tissue that enters the optic cup via the choroidal fissure gives increase to the hyaloid vessels during intrauterine life and also forms a network of delicate fibers between the lens and the retina, which later fills with a transparent gelatinous substance forming the vitreous body. The hyaloid artery provides the blood supply to the developing lens as well as the retina, and is a branch of the ophthalmic artery which reaches the inner chamber of the optic globe via the choroidal fissure on the ventral surface of the optic cup and the optic stalk. The portion of the hyaloid artery that traverses the vitreous region to the lens degenerates in fetal life as the lens matures. As the hyaloid vessels in this segment are obliterated, a hyaloid canal is left behind. The remainder of this artery remains as the central artery of the retina as the lips of the choroidal (retinal) fissure fuse during the sixth and the seventh week ( Fig. 2 ).
Development of the Other Orbital Structures
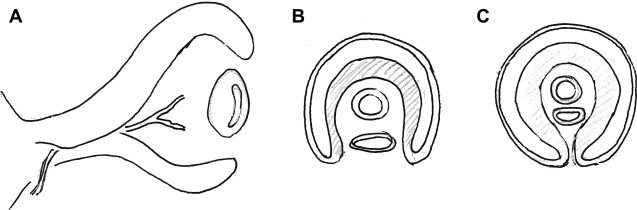
The extraocular muscles develop from the mesenchyme surrounding the optic vesicle. It is believed that the connective tissue associated with the extrinsic ocular muscles is derived from the neural crest. The primordia of the first of the extraocular muscles appear as early as day 28, and the primordia of all muscles appear by the sixth week.
The oculomotor nerve is the first to develop followed by the trochlear and the abducent nerves appear during the fifth week of the fetal life, on days 32 and 33.
Eyelid grooves first appear on day 37. Small folds of the surface ectoderm with a mesenchymal core appear on the cranial and caudal aspects of the developing cornea, representing the primordia of the upper and lower eyelids during the seventh week. These folds grow toward each other and fuse. The space between the fused eyelids and cornea develops into the conjunctival sac. The eyelids separate between the fifth and seventh months of development.
The neurocranium (part of the head that houses the brain) is divided into 2 parts – the chondrocranium made up of bones that ossify in cartilage forming the structures of the skull base, and the desmocranium consisting of the flat bones that form the calvarial vault and ossify in membrane. The body of the sphenoid develops from the hypophyseal cartilages of the prechordal chondrocranium, with the lesser and greater wings developing from the adjacent mesenchymal condensations called the ala orbitalis and the ala temporalis respectively. The mesenchyme for the development of the face including the nasal and lacrimal bones is derived from the neural crest cells. The ossification of these bones starts between the sixth and 16th week. In the fetus, the neurocranium far exceeds the size of the viscerocranium (facial skeleton), and the fetal orbit is represented mainly by the frontal bone.
In the 1970s, working on a doctoral thesis, deHaan described the development of the orbit in great detail, although the thesis remained incomplete at the time. In the embryonic and early fetal stages, the primordium of the eye is present; however, the other tissues of the eye socket cannot be identified. Only vague condensations of the mesenchymal connective tissue are seen. The author refers to the region as the primordial orbit during the early part of the fourth month (days 95–105 after ovulation) when the connective tissue structures can be clearly distinguished. The face of the fetus acquires more human features during the third month as the eyes, which are oriented laterally until then, move to the anterior aspect of the face. The optic nerves are at an angle of 180 degrees in embryonic stage 16 (day 37), 84 degrees in stage 23 (day 57), and 55 degrees from a fetus of 65 mm crown breech length (CBL) (third month) until term. Further maturation and rotation of the axis of the ocular globe occurs until adulthood.
Genetic Factors in the Early Development of the Eye
The PAX6 gene is considered the master regulator or master selector of eye development. This transcription factor is expressed in the anterior neural ridge of the neural plate before neurulation begins. At this stage, a single eye field exists that later separates into 2 optic primordia under the influence of the sonic hedgehog (SHH) expressed in the prechordal plate, by upregulation of the PAX2 gene and downregulation of PAX6 gene in the center of the single eye field. Later in development, the PAX2 gene is expressed in the optic stalks, whereas the PAX6 gene is expressed in the optic cup and the overlying surface ectoderm where it is considered essential for the lens development. After the induction of the lens, bone morphometric protein (BMP) 7 is necessary to maintain eye development. Other genes that have been associated with early eye development include the SOX2 gene (expressed in the optic cup along with PAX6), the OTX2 gene, and the fibroblast growth factor (FGF), signals from which overlap with the BMP7 gene along with other members of the BMP family and the retinal homeodomain transcription factor (Rx/RAX).
Congenital/developmental malformations of the ocular globe
Anomalies of Number and Size
Anophthalmia and microphthalmia
Anophthalmia and microphthalmia are among the most common ocular birth defects and a significant cause of congenital blindness. Either of these defects can occur in isolation or as part of a syndrome. Anophthalmia (or anophthalmos) refers to the absence of the ocular globe, whereas microphthalmia refers to a small size of the ocular globe. The mean maximum axial length of the neonatal human eye is approximately 17 mm (compared with 23.8 mm of the adult eye).
Primary anophthalmia is a sporadic anomaly unassociated with a systemic defect, where the ocular primordia never form, with failure of outpouching of the wall of the forebrain. Seventy-five percent of the cases may be bilateral. The defect occurs early in development, at about 22 to 27 gestational days (3 mm embryo stage). Secondary anophthalmia results from degeneration or failure of development of the entire neural tube. Consecutive anophthalmos refers to degeneration of the optic vesicle after evagination and, consequently, some neuroectodermal elements may be present. There have been observations of extraocular muscles inserting into a fibrous nodule in an apparently or clinically anophthalmic socket that might represent an aborted eye ( Fig. 3 ). A similar situation may be inferred if portion of the optic nerve or chiasm is formed. The size of the orbit and the conjunctival sac are reduced in anophthalmia. Disorganized mesodermal and surface ectodermal elements may be present, although neuroectodermal elements are absent in true anophthalmia. However, this is often difficult to confirm, and many cases may represent a severe microphthalmia.
Microphthalmia can be unilateral or bilateral, and may represent a primary ocular developmental abnormality, be associated with a craniofacial dysplasia, or be part of larger syndrome, such as with chromosomal anomalies including trisomy 13 and trisomy 18. It can also occur with other ocular processes including congenital infections (rubella), septo-optic dysplasia, or retinopathy of prematurity. Most common ocular malformations associated with microphthalmia include persistent hyperplastic primary vitreous, nuclear cataract, and coloboma.
Several genetic mutations have been linked to anophthalmia and microphthalmia. The first gene to be identified was the PAX6 gene, although SOX2 is a major causative gene. Other reported genetic associations include RAX gene leading to anophthalmia in humans, abnormalities of SOX2 gene, and loss of function mutation in the OTX2 and CHX10 genes related to microphthalmia.
Cyclops and synophthalmia
Both refer to a clinical state in which only 1 eye is present. Cyclops, or cyclophthalmia, refers to a complete fusion or a single median eye that is associated with holoprosencephaly. Synophthalmia represents partial fusion of optic vesicles, resulting in duplication of some anterior structures.
Cryptophthalmos
As discussed earlier in the article, the eyelid folds appear during the seventh week, grow toward each other, and fuse. The eyelids later separate between the fifth and seventh months of development. Cryptophthalmos is a result of failure of development of the eyelid folds. The eyelids, eyebrows, and the cornea are absent, and skin is continuous from the forehead to the cheeks. Absence of the eyelashes, meibomian glands, and the lacrimal apparatus is noted.
Cryptophthalmos is often a part of a systemic syndrome. Imaging studies are needed to show the underlying ocular globes and the other orbital structures before surgical intervention.
Buphthalmos
Buphthalmos (also called hydrophthalmos) refers to an enlarged eye caused by congenital or infantile glaucoma. It can be an isolated finding and bilateral in up to 80% of cases. The corneal diameter is increased. This may occur in conjunction with systemic disorders such as Marfan syndrome.
Enlargement of ocular globe from buphthalmos must be differentiated from other conditions resulting in uniform or focal enlargement of the eye. In buphthalmos, the globe is generally enlarged uniformly, but it may occasionally have oval or bizarre configurations. Imaging is helpful in excluding an underlying ocular mass such as retinoblastoma. In the absence of an intraocular mass, an enlarged ocular globe occurring early in life may also be associated with neurofibromatosis type I or Sturge-Weber syndrome.
An enlarged ocular globe can also be a feature of axial myopia with an elongated anteroposterior dimension, but with a normal cornea. In cases of severe myopia, a posterior outpouching/ posterior staphyloma may be present, occurring as a result of thinning of the posterior sclera.
Congenital Cystic Eye
This is a rare anomaly that results from the failure of invagination of the optic vesicle during embryogenesis. This condition presents at birth as a complex cyst lined by cells derived from undifferentiated retina and retinal pigment epithelium, or may present clinically following postnatal expansion.
The neuroglial tissue consists of dystrophic calcified bodies and degenerated primitive nerve fibers. The cyst may enlarge because of the fluid produced by glial tissue. The main differential considerations for cystic anomalies include microphthalmia with cyst, microphthalmia with cystic teratoma, ectopic brain tissue, and meningoencephalocele.
Coloboma
This is a developmental abnormality that occurs as a result of failure of closure of the embryonic choroidal fissure, and presents as a cleft in the inferonasal quadrant. Depending on the extent of involvement, colobomas may affect the iris, ciliary body, retina, choroid, and sclera, and may even involve the optic nerve. The developmental insult occurs during the period of gestational days 35 to 41. Colobomas can be seen in isolation or with other ocular disorders, or with multisystem abnormalities.
Colobomas may be present with normal or with small size of the ocular globes and are often bilateral. Sometimes there may be a mild abnormality with unremarkable findings on imaging. In the mildest form, an optic nerve coloboma may present as a visually insignificant enlargement of the optic disc with a large optic cup. The globe may be misshapen, with a very deep optic cup, extending along the optic nerve ( Fig. 4 ). In the morning glory syndrome, the disc is posteriorly displaced in a posterior staphylomatous excavation at the optic nerve head. The retina in the coloboma is at risk for spontaneous detachment.
A more severe abnormality is microphthalmia associated with a colobomatous cyst. The relative sizes of the ocular globe and the cyst are variable and, on occasion, the cyst may be much larger than the ocular globe ( Fig. 5 ). In these cases, the defect in the sclera allows for an extraocular herniation of the intraocular neural ectoderm and the vitreous body to form a cyst with a tunnel-like connection to the globe. The ocular globe may be distinguished from the cyst by the presence of the other intraocular contents, such as the lens.
Colobomas can be an isolated abnormality in autosomal dominant coloboma-microphthalmos. Colobomas can occur in several multisystem syndromes, the most notable being the CHARGE (coloboma, heart defects, choanal atresia, retarded growth and development, genital malformations and ear anomalies) association. Genetic disorders and associations include focal dermal hypoplasia, Aicardi syndrome, brachi-oculo-facial syndrome, and trisomies 13 and 18, and have also been reported with fetal alcohol syndrome.
Persistent Hyperplastic Primary Vitreous
Persistent hyperplastic primary vitreous (PHPV) is a congenital, usually unilateral, abnormality clinically characterized by leukokoria in a microphthalmic eye. This condition results if the portion of the hyaloid artery that traverses the vitreous region from the optic nerve head to the lens and the fibrovascular tissue of the primary vitreous fail to degenerate and resorb, with a resultant vascularized plaque on the posterior aspect of the ocular lens. The term persistent fetal circulation has also been suggested for this entity.
There is generalized increased density of the vitreous on computed tomography (CT), which may show enhancement following contrast administration. Microphthalmia and visualization of portions of a septum extending from the optic nerve head to the site of the primary vitreous behind the lens is diagnostic. Tubular, cylindrical, triangular, or other intravitreal densities suggest persistence of fetal tissue in the Cloquet canal or congenital nonattachment of the retina. There may be a generalized increase in the density of the vitreous chamber, but calcification is absent, allowing differentiation from retinoblastoma. Similarly, magnetic resonance (MR) imaging may show abnormal hyperintensity of the vitreous related to the blood products. The lens may also appear abnormal and MR imaging may show abnormal enhancement in the fibrovascular mass posterior to the lens ( Fig. 6 ).
Bilateral PHPV may be present in a congenital syndrome such as Warburg disease, and similar bilateral findings of a retrolental mass and hyperdense retinal detachment have been described with an X-linked recessive condition called Norrie disease (oculoacoustic cerebral degeneration) ( Fig. 7 ). When microphthalmia is present, it may also aid differentiation from Coats disease and retinoblastoma, although differentiation of PHPV from a noncalcified retinoblastoma in a normal-sized globe may be difficult. The other clinical condition in the differential diagnosis of leukokoria associated with microphthalmia in the neonatal age group is retinopathy of prematurity. Many of these conditions eventually lead to phthisis bulbi and may show calcification.
Retinopathy of Prematurity
Retinopathy of prematurity (ROP: previously called retrolental fibroplasia) is a condition seen in premature infants who have required prolonged oxygen therapy for respiratory distress syndrome/hyaline membrane disease. This is a condition limited to the immature retinal vasculature, and the involvement is usually bilateral. There is vasoconstriction and chronic retinal ischemia that induces abnormal vascular proliferation and neovascularization of the retina that extends into the vitreous. Vitreous hemorrhage and tractional retinal detachment may occur. CT and MR findings are relevant to the stage of blood products. Imaging is usually not performed. At an early stage, there may be no specific signs on CT or MR imaging, except that the eye may be micro-ophthalmic. In advanced cases, the differential considerations include PHPV, retinoblastoma, endophthalmitis, and other conditions that result in retinal detachment. Calcification is rarely present in ROP, helping differentiation from retinoblastoma.
Coats’ Disease
This is an idiopathic primary retinal vascular disease, characterized by telangiectasias, neovascularization, and beading and tortuosity of retinal vessels, with progressive accumulation of lipoproteinaceous exudate leading to retinal detachment. Although this is a developmental abnormality, patients (more frequently boys) often present later in childhood, between 4 and 6 years of age (rarely younger than 2 years), when the main differential diagnosis is retinoblastoma. Both can present with a triad of retinal detachment, dilated retinal vessels, and appearance of a subretinal mass in a child presenting with leukokoria.
On CT, advanced Coats disease shows abnormal density posterior to a contracted vitreous, with infrequently reported calcification on pathologic specimens. There is no enhancement of the subretinal mass (unlike retinoblastoma), and there may be a slight linear enhancement at the boundary between the mass and the vitreous. The abnormal tissue is intraocular (unlike retinoblastoma, in which extraocular spread is possible) and the eye is normal sized to slightly enlarged. On MR imaging, the subretinal material appears hyperintense on T1-weighted and T2-weighted images, although the T2 signal can be varied because of areas of fibrosis and organized hemorrhage.
Phakomatoses
As mentioned earlier, macrophthalmos (large eye) may be associated with neurofibromatosis type I as well as Sturge-Weber syndrome. Hamartomas called Lisch nodules are known to occur in the iris in neurofibromatosis, but are too small to be seen on imaging.
In von Hippel-Lindau (VHL) disease, retinal angiomatosis or capillary hemangioblastomas are characteristic lesions that are often bilateral and multifocal, being the first manifestation in about 50% of patients with VHL. The diagnosis is made on ophthalmoscopic examination, with a minor role for radiology, because most retinal lesions are too small. MR imaging may show signal abnormalities in the globe related to retinal detachment and rarely show the enhancing ocular lesions. The patients are typically in the third decade of life.
In tuberous sclerosis, retinal hamartomas may occur that appear as smooth elevations that may calcify. Retinal giant cell astrocytomas have been reported.
Retinal arteriovenous malformations are known to occur in the Wyburn-Mason syndrome, along with lesions in the ipsilateral brain ( Fig. 8 ) and, less frequently, the face.
Related posts:
 Demystifying Congenital Malformations for Imaging Specialists
Demystifying Congenital Malformations for Imaging Specialists
 Development and Dysgenesis of the Cerebral Cortex: Malformations of Cortical Development
Development and Dysgenesis of the Cerebral Cortex: Malformations of Cortical Development
 Imaging of Congenital Spine and Spinal Cord Malformations
Imaging of Congenital Spine and Spinal Cord Malformations
 Neurosurgical Management of Congenital Malformations and Inherited Disease of the Spine
Neurosurgical Management of Congenital Malformations and Inherited Disease of the Spine
 Congenital Malformations of the Temporal Bone
Congenital Malformations of the Temporal Bone
 Hemangiomas and Vascular Malformations of the Head and Neck: A Simplified Approach
Hemangiomas and Vascular Malformations of the Head and Neck: A Simplified Approach
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


