There are a wide variety of congenital midface abnormalities that originate during transformation of the first pair of pharyngeal arches into adult structures. Computed tomography and magnetic resonance imaging are important components in the comprehensive evaluation of these lesions. A detailed understanding of midface embryogenesis and developmental anatomy is important in directing appropriate patient management.
Development of the face
Congenital anomalies of the midface originate during transformation of the first pair of pharyngeal arches into adult structures. The first stage of facial development occurs during the fourth week of gestation around the primordial mouth or stomodeum. The first pair of pharyngeal arches gives rise to 5 facial prominences: a single frontonasal prominence, paired maxillary prominences, and paired mandibular prominences. These 5 facial prominences form the boundaries of the stomodeum, as illustrated in Fig. 1 . The facial prominences are the result of migration and proliferation of neural crest cells into the first pair of pharyngeal arches. Neural crest cells are the predominant source of facial connective tissue components including bone, cartilage, and ligaments.
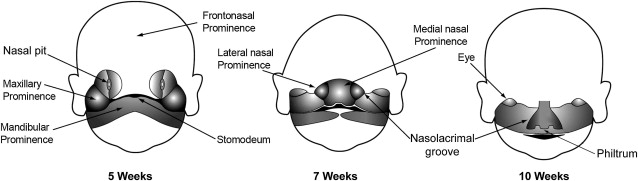
Facial development mainly takes place between the fourth and eighth week of gestation. The frontal part of the frontonasal prominence forms the forehead and dorsum of the nose. The nasal part of the frontonasal prominence gives rise to bilateral nasal placodes. A nasal placode is composed of an oval thickening of surface ectoderm. The margins of a nasal placode proliferate to form a ridge with a medial and lateral nasal prominence. The central depression in a nasal placode between the nasal prominences is the nasal pit. The developing nasal pits deepen to become the anterior nares (nostrils), and nasal cavity.
The lateral nasal prominences are separated from the maxillary prominences by clefts called the nasolacrimal grooves. Medial growth of the maxillary prominences causes the medial nasal prominences to move toward each other and fuse. The fused medial nasal prominences give rise to the medial upper lip, philtrum (infranasal depression), primary palate, incisor teeth, and nasal septum. The lateral nasal prominences form the nasal alae (sides of the nose). The maxillary prominences form the lateral upper lip, most of the maxilla, and secondary palate.
Choanal atresia and stenosis
The developing nasal pits deepen to form the primitive nasal cavity. At first, the posterior aspect of the nasal cavity is separated from the oral cavity by the oronasal membrane. In the sixth week of gestation, the oronasal membrane normally ruptures. The site of continuity between the posterior nasal and oral cavities are the choanae. Later, after the development of the secondary palate, the choanae are located between the nasal cavity and nasopharynx. Cells that line the nasal cavity proliferate to form a temporary epithelial plug that is subsequently resorbed.
Choanal atresia is the most common cause of neonatal nasal obstruction, occurring in up to 1 in 5000 newborns. Bilateral choanal atresia causes respiratory distress in newborns who are obligate nose breathers. The diagnosis can be suggested by the inability to advance a nasal catheter. Computed tomography (CT) examination is the imaging modality of choice in neonates with suspected choanal atresia. CT should be performed immediately after suctioning of nasal secretions. It can identify whether the obstruction is osseous (85%) or membranous (15%). Osseous choanal atresia is related to failure of rupture of the oronasal membrane. Membranous choanal atresia is caused by incomplete resorption of the nasal epithelial plugs.
Choanal atresia is commonly associated with other congenital anomalies (50%–70%). Common associations include coloboma, heart defects, choanal atresia, retarded growth and development, genital malformations and ear anomalies (CHARGE) syndrome, Apert syndrome, Crouzon syndrome, Treacher Collins syndrome, and bowel malrotation. Individuals with CHARGE syndrome have various combinations of the abnormalities listed in the acronym, as shown in Figs. 2 and 3 .
CT examination may show a bony or soft tissue septum extending across the posterior choanae. Associated imaging features in bony choanal atresia include inward bowing of the posterior maxilla, and fusion or thickening of the vomer. In children younger than 2 years old, narrowing of the posterior choanal opening less than 0.34 cm is defined as choanal stenosis. The current treatment of choanal atresia causing neonatal nasal obstruction is endoscopic perforation or choanal reconstruction.
Choanal atresia and stenosis
The developing nasal pits deepen to form the primitive nasal cavity. At first, the posterior aspect of the nasal cavity is separated from the oral cavity by the oronasal membrane. In the sixth week of gestation, the oronasal membrane normally ruptures. The site of continuity between the posterior nasal and oral cavities are the choanae. Later, after the development of the secondary palate, the choanae are located between the nasal cavity and nasopharynx. Cells that line the nasal cavity proliferate to form a temporary epithelial plug that is subsequently resorbed.
Choanal atresia is the most common cause of neonatal nasal obstruction, occurring in up to 1 in 5000 newborns. Bilateral choanal atresia causes respiratory distress in newborns who are obligate nose breathers. The diagnosis can be suggested by the inability to advance a nasal catheter. Computed tomography (CT) examination is the imaging modality of choice in neonates with suspected choanal atresia. CT should be performed immediately after suctioning of nasal secretions. It can identify whether the obstruction is osseous (85%) or membranous (15%). Osseous choanal atresia is related to failure of rupture of the oronasal membrane. Membranous choanal atresia is caused by incomplete resorption of the nasal epithelial plugs.
Choanal atresia is commonly associated with other congenital anomalies (50%–70%). Common associations include coloboma, heart defects, choanal atresia, retarded growth and development, genital malformations and ear anomalies (CHARGE) syndrome, Apert syndrome, Crouzon syndrome, Treacher Collins syndrome, and bowel malrotation. Individuals with CHARGE syndrome have various combinations of the abnormalities listed in the acronym, as shown in Figs. 2 and 3 .
CT examination may show a bony or soft tissue septum extending across the posterior choanae. Associated imaging features in bony choanal atresia include inward bowing of the posterior maxilla, and fusion or thickening of the vomer. In children younger than 2 years old, narrowing of the posterior choanal opening less than 0.34 cm is defined as choanal stenosis. The current treatment of choanal atresia causing neonatal nasal obstruction is endoscopic perforation or choanal reconstruction.
Congenital nasal piriform aperture stenosis
Congenital nasal piriform aperture stenosis is an infrequent cause of nasal obstruction in neonates and infants. It can mimic choanal atresia, including the inability to easily advance a nasal catheter. This condition is exacerbated by upper respiratory tract infections that further compromise the narrow nasal aperture.
CT examination is the imaging modality of choice to accurately measure the piriform aperture width. A width less than 11 mm in a term infant is diagnostic. Associated imaging features include an abnormal dentition with a central megaincisor, and a triangular configuration of the palate ( Fig. 4 ). The underside of the palate often has a midline bone ridge. Magnetic resonance (MR) imaging is helpful to evaluate for associated intracranial anomalies including holoprosencephaly and pituitary abnormalities.
One theory about the pathogenesis of nasal piriform aperture stenosis is faulty development of the palate. The palate is the floor of the nasal cavity and roof of the mouth. It develops from 3 structures: the primary palate and 2 lateral palatal shelves. The primary palate constitutes the small part of the adult hard palate anterior to the incisive fossa. The 2 lateral palatal shelves form the remainder of the adult hard palate and soft palate. Normally, the primary palate is formed by the fused medial nasal prominences in the sixth week of gestation. The secondary palate develops as 2 lateral palatal shelves extend from the inner maxillary prominences. Following a sequence of intricately timed events, the 2 lateral palatal shelves fuse to each other, the primary palate, and nasal septum.
Nasal piriform aperture stenosis can be explained by a deficiency of the fused medial nasal prominences, with subsequent development of a small triangular primary palate, abnormal incisors, and narrow anterior nasal cavity. The midline bony ridge along the underside of the palate may be related to abnormal overlap and adhesive contact of the palatal shelves during formation of the secondary palate. Most patients with congenital nasal piriform aperture stenosis have an excellent prognosis. These patients are usually treated conservatively with special feeding techniques until the obstruction is relieved by growth of the nasal cavity. Severe cases may require surgical reconstruction.
Cleft lip, cleft palate, and facial clefts
Cleft lip is the most common craniofacial malformation. The incidence of cleft lip varies between ethnic groups (white population 1:1000 live births). It results from failure of fusion of the maxillary prominence on the affected side with the medial nasal prominence. The least severe form of a cleft lip involves only the superficial vermilion border of the lip. Increasingly severe clefts extend through the maxillary alveolar arch between the lateral incisor and cuspid tooth. If epithelium is entrapped between the globular portion of the medial nasal process and the maxillary process, a globulomaxillary (fissural) cyst can develop between the maxillary lateral incisor and cuspid. Cleft lip may occur with or without a cleft palate. Cleft lip is bilateral in 20% of cases. Prenatal ultrasound can reliably show a cleft lip as an abnormal hypoechoic line extending from the upper lip into the nostril.
Cleft palate is caused by incomplete fusion of the lateral palatal shelves with each other, the nasal septum, and the primary palate. It can be unilateral or bilateral. The least severe form of a cleft palate is a bifid uvula. More severe clefts extend anteriorly along the expected lines of fusion of the lateral palatal shelves and posterior aspect of the primary palate. The nasal septum may fuse with the left or right palatal shelf or neither. An isolated cleft palate may be missed on prenatal ultrasound. In the setting of bilateral cleft lip and palate, the infranasal soft tissue, alveolar ridge, and dental structures between the clefts can cause a conspicuous premaxillary protrusion.
The development of the face may be disrupted by multiple toxic substances, nutritional imbalance, and genetic factors. There are several different classifications used to describe rare facial clefts, including the Tessier, DeMeyer, and Sedano systems. The Tessier system numbers the topographic location of facial clefts from 1 to 14. Clefts 1 to 7 are below the orbit, and 8 to 14 are above. Rare facial clefts can be predicted based on the expected embryologic fusion lines of the 5 facial prominences ( Fig. 5 ). For example, incomplete fusion of the paired mandibular prominences leads to a midline mandibular cleft. Failure to merge a maxillary and lateral nasal prominence causes an oblique facial cleft (orbitofacial fissure) extending from the nasal ala to the medial canthus. Within the oblique facial cleft, the nasolacrimal duct remains as an open groove. Facial clefts that do not respect embryologic fusion lines are most likely secondary to amniotic band syndrome.
Rare facial clefts with hypotelorism or hypertelorism are more likely to have coexisting brain anomalies. Facial clefts with hypotelorism have been described with ventral induction malformations ranging from alobar holoprosencephaly to septo-optic dysplasia. Facial clefts with hypertelorism can have associated dysgenesis of the corpus callosum, and pituitary gland anomalies.
Nasolacrimal duct stenosis and atresia
The nasolacrimal duct develops from an ectodermal thickening in the floor of the nasolacrimal groove at 1 month gestational age. This ectodermal thickening makes a solid cord that separates from the surface ectoderm and is enveloped by the underlying mesenchyme. The solid cord becomes canalized to form the nasolacrimal duct. The superior end of the nasolacrimal duct expands to create a lacrimal sac. The inferior end of the duct empties into the inferior meatus via the Hasner membrane. Spontaneous rupture of the Hasner membrane with production of a mucosal fold called the Hasner valve is required for nasolacrimal duct patency. Rupture of the Hasner membrane likely occurs after birth when a newborn first attempts to breathe and cry.
Nasolacrimal duct stenosis is common in neonates. It is caused by partial persistence of the Hasner membrane. CT is the imaging modality of choice in suspected nasolacrimal duct anomalies. Imaging findings can include an asymmetrically enlarged opacified nasolacrimal canal ( Fig. 6 ). However, nasolacrimal duct stenosis can also appear normal. Nasolacrimal stenosis resolves spontaneously in 90% of infants within the first year of life with conservative management. Prophylactic antibiotics may be administered during this period to prevent periorbital cellulitis. In selected cases, canalicular probing and intubation are widely accepted treatment options.
Nasolacrimal duct atresia is an uncommon congenital anomaly. It is caused by failure to canalize the epithelial cord that is the precursor of the nasolacrimal duct. Usually atresia of the nasolacrimal duct occurs at its inferior aspect. Congenital atresia of the lacrimal drainage system can also occur superiorly at the lacrimal puncta and canaliculi.
Nasolabial cysts are briefly described in this article because their origin is most likely related to aberrant nasolacrimal duct development. If an embryonic nasolacrimal epithelial rest becomes entrapped within the nasal alar region, it can form a benign cystic lesion called a nasolabial cyst. These rare cysts may encroach on the nasal aperture. Their characteristic extraosseous location should prevent confusion with maxillary cysts.
Dacryocystoceles
Dacryocystoceles are a frequent cause of neonatal nasal obstruction. A dacryocystocele is caused by obstruction of the lacrimal drainage system above and below the lacrimal sac. Subsequently the lacrimal sac becomes distended with accumulated secretions and prone to infection. The distal obstruction is often an imperforate Hasner membrane. The proximal obstruction is most likely at the Rosenmuller valve near the entrance of the common canaliculus into the lacrimal sac. CT is the imaging modality of choice to diagnose a dacryocystocele and exclude other nasal masses. The imaging features include a round cystic mass at the medial canthus with a thin peripheral wall ( Fig. 7 ). Associated proximal nasolacrimal duct dilatation with bony remodeling can produce elevation of the adjacent inferior turbinate and contralateral shift of the nasal septum.
Dacryocystoceles require expedient management to decrease the risk of superimposed infection and scarring of the lacrimal drainage system. Treatment ranges from manual pressure on the lacrimal sac to minimally invasive probing and irrigation of the canaliculi. Endoscopic resection and marsupialization are reserved for severe cases. Infection of an obstructed lacrimal sac is called dacryocystitis. It is best evaluated with a contrast-enhanced CT examination. Imaging features include a distended lacrimal sac with associated enhancing thickened walls, and surrounding preseptal soft tissue swelling ( Fig. 8 ).
Related posts:
 Demystifying Congenital Malformations for Imaging Specialists
Demystifying Congenital Malformations for Imaging Specialists
 Development and Dysgenesis of the Cerebral Cortex: Malformations of Cortical Development
Development and Dysgenesis of the Cerebral Cortex: Malformations of Cortical Development
 Imaging of Congenital Spine and Spinal Cord Malformations
Imaging of Congenital Spine and Spinal Cord Malformations
 Neurosurgical Management of Congenital Malformations and Inherited Disease of the Spine
Neurosurgical Management of Congenital Malformations and Inherited Disease of the Spine
 Congenital Malformations of the Temporal Bone
Congenital Malformations of the Temporal Bone
 Hemangiomas and Vascular Malformations of the Head and Neck: A Simplified Approach
Hemangiomas and Vascular Malformations of the Head and Neck: A Simplified Approach
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


