Abstract
Cornelia de Lange syndrome (CdLS) should be considered in any fetus with growth restriction in addition to limb or craniofacial abnormalities, with or without a congenital heart defect. Classic sonographic signs of CdLS include increased first-trimester nuchal translucency, intrauterine growth restriction, short or absent upper limbs, absent digits, fifth finger clinodactyly, syndactyly, radial hypoplasia, microcephaly, micrognathia, and congenital heart defects. However, unless there is a previous family history of CdLS, prenatal diagnosis is unlikely to be possible, and the diagnosis will have to be made after postnatal examination. In families with a history of CdLS where a mutation has been identified, prenatal diagnosis by chorionic villus sampling or amniocentesis is likely to be available. Normal first-trimester nuchal translucency, normal first-trimester or second-trimester maternal serum pregnancy-associated plasma protein A, or a normal second-trimester ultrasound examination in a fetus with a family history of CdLS is largely reassuring but cannot entirely rule out recurrence. Because most cases of CdLS are de novo , couples who do not have CdLS but have a previously affected child have a 1.5% risk of recurrence. If a parent of the affected child has CdLS, there is a 50% chance of a recurrence for each pregnancy.
Key Words
microbrachycephaly, upper limb reduction defects, oligodactyly, congenital heart defects
Introduction
Cornelia de Lange syndrome (CdLS), also known as Brachmann-de Lange or de Lange syndrome, is a condition characterized by mental retardation, craniofacial dysmorphism, prenatal and postnatal growth failure, hirsutism, and upper limb abnormalities. Other defects occasionally associated with CdLS include congenital cardiac defects and genital anomalies. There are three subtypes of CdLS. CdLS1, the classic form, is caused by autosomal dominant mutations in the NIPBL gene; 99% are de novo , whereas 1% are attributed to parental germline mosaicism. CdLS2 is caused by mutations in the X-linked SMC1A gene. CdLS3, a mild variant of the disorder, is caused by mutations in the SMC3 gene. In individuals with CdLS3, the facial dysmorphisms, anatomic anomalies, and learning deficits can be minor or completely absent. The number of individuals with mild CdLS or CdLS3 is thought to be underreported.
Disease
Definition
The diagnosis of CdLS is clinical; gene testing is available to support the clinical diagnosis, but not all individuals with CdLS will have a known genetic mutation. Individuals affected with classic CdLS have distinct craniofacial abnormalities including high arched palate, microbrachycephaly, and micrognathia. In addition, hirsutism, including synophrys, hirsute ears, and long, coarse eyelashes are noted in most individuals. Prenatal growth failure continuing throughout life is identified in greater than 95% of affected individuals, as are severe to profound mental retardation and upper limb abnormalities. Genetic testing can be helpful in determining which subtype of CdLS the individual has, the cause of the syndrome, and the risk to other family members.
Prevalence and Epidemiology
The incidence of classic CdLS is 1 : 100,000 to 1 : 10,000. It is thought that a large proportion of individuals with mild CdLS are not diagnosed, and the prevalence may be much higher than these estimates suggest. CdLS affects all ethnic groups and can be found equally in males and females.
Etiology and Pathophysiology
Several genes have been associated with CdLS. Approximately 60% of individuals with this condition have been found to have heterozygous alterations in the NIPBL gene, another 5% of individuals with CdLS have been found to have mutations in the SMC1A gene, 4% have mutations within the HDAC8 gene, 1%–2% have mutations in the SMC3 gene, and <1% have alterations within the RAD21 gene. No genetic abnormalities are identified in approximately 30% of affected individuals. For “mutation-negative” patients with CdLS, deeper sequencing from multiple tissues may lend a genetic diagnosis.
NIPBL mutations are associated with the CdLS1subtype. Although the exact function of the NIPBL protein, delangin, is unknown, it is thought that it is important in proper sister chromatid cohesion. Individuals with CdLS1 may display the classic signs of this syndrome or may display milder manifestations, although most individuals with more severe manifestations of CdLS have mutations in the NIPBL gene. In most cases, these NIPBL gene alterations are de novo , although autosomal dominant transmission has been reported in several families.
Mutations in the SMC1A and SMC3 genes have been identified in only a few affected individuals, and genotype-phenotype associations are not well established, although individuals with mutations in these genes appear to have milder manifestations of the syndrome than individuals with NIPBL mutations. The limb and other structural defects seen in individuals with CdLS are rarely found in individuals with mutations in these genes. The SMC1A and SMC3 proteins are components of the cohesin complex. Similar to the NIPBL protein, they are thought to be important in cohesion of sister chromatids. For this reason, CdLS is thought to be a cohesinopathy. SMC1A mutations can cause CdLS in males or females. CdLS2 is thought to be X-linked dominant. SMC3 mutations are associated with the CdLS3 subtype and are typically de novo but can be associated with autosomal dominant transmission.
Manifestations of Disease
Clinical Presentation
All three subtypes and CdLS resulting from unknown genetic cause can manifest similarly; however, manifestations are often milder in individuals with no mutation or with SMC1A or SMC3 mutations than in individuals with NIPBL mutations. Limb and other structural defects are typically absent in individuals with SMC1A or SMC3 mutations. Common manifestations of this condition include the following:
- •
prenatal and postnatal growth restriction
- •
intellectual disability with mean intellectual quotient of 53
- •
autistic features
- •
craniofacial abnormalities
- •
microbrachycephaly
- •
synophrys
- •
high arched palate
- •
micrognathia
- •
flat, broad nasal bridge with anteverted nares
- •
- •
limb anomalies
- •
limited elbow extension
- •
upper limb reduction defects
- •
radial hypoplasia
- •
proximally placed thumbs
- •
fifth finger clinodactyly
- •
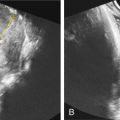
oligodactyly ( Fig. 127.1 )

Fig. 127.1
Oligodactyly.
- •
syndactyly
- •
- •
hirsutism
- •
sensorineural or conductive hearing loss
- •
myopia
- •
late-erupting, widely spaced teeth
- •
short neck
- •
congenital heart defects in 30% of patients
- •
other structural birth defects (cleft lip/palate, congenital diaphragmatic hernia [ Fig. 127.2 ])