DEVELOPMENTAL ABNORMALITIES OF THE INNER EAR AND CRANIAL NERVE VIII
KEY POINTS
- Computed tomography is critical to medical decision making in developmental abnormalities of the inner ear and cranial nerve VIII.
- Reports must be logically constructed and comprehensive, anticipating all anatomic and pathologic abnormalities that might lead to altered treatment plans.
Development of the inner ear from the primitive otocyst is separate from that of the middle and external ear, which come from the first and second branchial arches. The structures involved in each phase of development should be evaluated in the clinical context. Patients suspected of having congenital hearing deficits will usually present in distinct groups: sensorineural hearing loss and potential inner ear anomalies, the subject of this chapter, and those within the spectrum of aural atresia or with an otherwise unexplained conductive hearing loss (Chapter 105). A combined inner and external ear anomaly reflects injury to the otocyst and the branchial apparatus usually requiring a chromosomal insult, a syndromic relationship (Chapter 107), or a severe multicentric and early acquired process such as rubella infection during gestation.
The written report of findings in anomalies of the temporal bone and cochleovestibular nerve (cranial nerve VIII) should reflect the developmental background of these anomalies. Such a report must include systematic documentation of the entire auditory pathway as seen on either computed tomography (CT) or magnetic resonance imaging (MRI), and a specific statement for each structure in this linkage should appear in every report. A reasonable approach to the general report format is to trace the sound, essentially vibrating air, from the outside to the brain stem and the facial nerve back out. For the purposes of discussion, the suggested report content can be relatively conveniently divided into three basic categories:
- Those related to external auditory canal (aural) atresia/middle ear anomalies discussed in Chapter 105
- Those related to possible inner ear and cochleovestibular nerve anomalous development discussed in this chapter
- Those that might impact cochlear implant candidates discussed in Chapter 136
For the patient with sensorineural hearing loss, the report should emphasize the sensorineural part of the pathway but must also consider the preceding conductive pathway. Facial nerve development is at the interface of these two functional units of hearing, but consideration of its anomalous development is mainly linked to disorders of the external canal and middle ear, where it can dramatically affect treatment options.
ANATOMIC AND DEVELOPMENTAL CONSIDERATIONS
Embryology
The ectodermally derived inner ear begins as the otic pit before becoming the primitive otocyst. The otocyst first forms the saccule and utricle and then sends out three projections. The anterior projection develops into the cochlear duct, the lateral projection becomes the lateral semicircular canal (SCC), and the posterosuperior projection becomes the superior and posterior SCCs. Cochlear development is complete at the 8th week of gestation. Formation of the saccule, endolymphatic duct, and utricle is completed within 11 weeks of gestation. The SCCs appear at the 6th week, and the superior SCC is completed by approximately 20 weeks, followed by the posterior SCC and finally the independently formed lateral SCC at about 22 weeks. The cochlear and vestibular aqueducts develop in conjunction with intracranial spaces, thereby completing the interconnection of the perilymphatic and endolymphatic spaces with the intracranial cerebrospinal fluid (CSF) spaces.
The various degrees of dysplasia of the cochlea and vestibular system observed with imaging are simply growth arrests of these otocyst projections at various stages of development. Depending on the time of growth, arrest malformations involve the cochlea, the cochlea in association with the vestibular system and vestibular aqueduct, or only the vestibular system. At the extreme is the Michel anomaly, a rare complete cochlear aplasia. At the other end of the spectrum from an imaging perspective are dysplasias limited to the membranous labyrinth that will have no or only indirect abnormal imaging evidence of their presence. The latter may in the future be more appropriately anticipated by genetic markers. In between these extremes are partial developmental errors named in the past with eponyms, such as Mondini malformation. Eponyms used to classify cochlear anomalies and those used to describe other inner ear anomalies are of historic interest but should now be dropped in favor of a logical description of the degree of maldevelopment as suggested by Jackler et al.1 over 20 years ago (Fig. 106.1). Those growth arrests can be reasonably linked to the embryologic stage of development and further associated with specific genetic errors. Such associations may help to anticipate and limit the damage of possibly associated genetic errors and, in the future, perhaps lead to some preventive strategies.
Applied Anatomy
A detailed and complete knowledge of normal temporal bone anatomy as presented in Chapter 104 is required for the evaluation of congenital anomalies. The most common key elements in this evaluation include the following:


FIGURE 106.1. Classification of inner ear malformations by developmental stage after the method proposed by Jackler. A: Cochlear dysplasia by type. B: Cochlear dysplasia by stage of development. C: Vestibular dysplasia by type.
- Soft tissues: Parotid gland and capsule attachments to the external auditory canal, mandible, and temporomandibular joint
- Temporal bone: External auditory canal, roof of the middle ear and mastoid, ossicles, inner ear, and internal auditory canal (IAC)
- Cochleovestibular nerve: Brain stem nuclei to points of end organ innervation
- Facial nerve: Brain stem to its stylomastoid foramen exit
- Vascular structures: Internal carotid artery, jugular vein and bulb, and major dural venous sinuses of the posterior fossa
IMAGING APPROACH
Techniques and Relevant Aspects
In general, CT and MRI for these developmental conditions always requires the highest possible spatial resolution, sometimes needing to balance that against required low-contrast resolution. CT and MRI are used in a complementary manner to evaluate patients with hearing loss. In purely sensorineural loss, MRI is frequently the more logical primary study.
The technical factors behind developing appropriate high-resolution data sets are discussed in detail in conjunction with general CT and MRI parameters (Chapters 1–3). In young children, an MRI examination will often require general anesthesia.
General imaging parameters used in this region of interest are presented in Appendixes A and B and are discussed in Chapter 104.
Pros and Cons
CT commonly is used when the critical information for medical decision making lies primarily in the bony anatomy––for example, the status of the ossicles and course of the facial nerve. It may also be used to screen for inner ear and eighth cranial nerve anomalies if one wishes to avoid the general anesthesia or heavy sedation usually required for a comprehensive MRI examination of the temporal bone in an infant, in most children under 8 years of age, or in patients who cannot cooperate for MRI or have some contraindication to its use.
MRI may be essential in complex cases when inner ear abnormalities may be associated with an abnormal status of the proximal seventh and eighth cranial nerves. MRI with high-resolution steady state images might suffice for both the anatomic assessment of the inner ear and cochleovestibular nerve in one test. It can also be used to demonstrate an unusually large endolymphatic sac. In case no inner ear or cochleovestibular nerve abnormalities are found, then additional MRI of the brain is useful to evaluate for a central origin of sensorineural hearing loss (SNHL) such as demyelination, migrational anomalies, or superficial siderosis.
Ultrasound is not used in this anatomic area of interest.
Catheter angiography has essentially no role, with magnetic resonance (MR) angiography and CT angiography capable of screening for vascular anatomy or anomalies whenever necessary.
Controversies
The utility of measurements of inner ear structures as a means of detecting subtle inner ear dysplasias is discussed in the Controversies section of Chapter 104.
In recent years, concerns have grown over the possibility that cochlear nerves may be hypoplastic and unable to support successful outcomes with cochlear implantation. Such a hypoplastic cochlear nerve has been identified in about 18% of hearing impaired children.2–5 It has yet to be established what constitutes a cochlear nerve size that will allow for success. CT may identify patients at risk for having a hypoplastic cochlear nerve using a small size of the bony cochlear nerve canal or aperture at the base of the modiolus as an indirect sign of a hypoplastic nerve.6
SPECIFIC DISEASE/CONDITION
Developmental Abnormalities of the Internal Auditory Canal, Inner Ear, and Cochleovestibular Nerve
Etiology
Aside from those due to known syndromes (Chapter 107), developmental abnormalities of the internal auditory canal, inner ear, and cochleovestibular nerve are likely due to spontaneous chromosomal abnormalities that may be linked to gestational infections or toxicity, but they usually have no truly identifiable cause.
Prevalence and Epidemiology
These anomalies are largely sporadic conditions but may be syndromic. About 1 in 1,000 children has significant hearing loss or is deaf at birth or in the prelingual period. Approximately 30% are syndromic and may present with conductive, sensorineural, or mixed hearing loss. The nonsyndromic forms are almost exclusively sensorineural. An additional 1 in 1,000 children becomes deaf before adulthood.
The most common demonstrable inner ear dysplasia that can be identified with modern imaging is the large vestibular aqueduct that is very commonly associated with an incomplete partition of the cochlea. This anomaly has been shown to be related to a recessive mutation in transported genes, which may occur spontaneously or as part of Pendred syndrome with the latter’s associated thyroid manifestations.7 Anomalies of the inner ear will be present on CT and/or MRI in about one third of patients with SNHL.8
Clinical Presentation
Many cases of inner ear dysplasia will be discovered soon after birth, as infants are routinely screened for hearing loss using otoacoustic emissions and auditory brain stem response testing. The former test may fail to identify children with abnormally developed or malfunctioning cochlear nerves and normally developed inner ears. Furthermore, children may develop severe to profound hearing loss during their first 2 years of life. The diagnosis of severe to profound hearing loss in early childhood is often made when the child misses normal speech development milestones.
Patients otherwise present with various degrees of hearing loss with at least some and usually a dominant sensorineural component. Hearing loss may be accompanied by vertigo and/or unsteadiness. Vestibular symptoms are usually progressive up to the point of very poor vestibular reflexes. Patients with an large vestibular aqueduct typically present with a history of progressive SNHL since infancy or childhood, often reported to be induced by minor trauma. Patients may also present with stigmata of aural atresia or syndromes known to affect the hearing pathways.
Pathophysiology and Patterns of Disease
The various degrees of dysplasia of the cochlea and vestibular system observed with imaging are simply growth arrests of these otocyst projections at various stages of development. The CT or MR images simply display the disordered development as dysplasia or aberrant development, hypoplasia or underdevelopment, or aplasia with no development. This fits well with logical classification systems established about 20 years ago for classifying these abnormalities.1


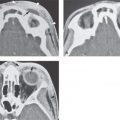
FIGURE 106.2. Cochleovestibular anomaly due to early failure in development in the third gestational week. On axial (A–C) and coronal (D–F) computed tomography images, two small cavities are present in place of the cochlea and vestibular system. This corresponds to labyrinthine aplasia and is a very rare condition in the past referred to as the Michel deformity.
Cochleovestibular Anomalies
Cochleovestibular abnormalities in their most gross forms and their related approximate times of growth arrest during gestation are shown in Figure 106.1 and in the images seen in Figures 106.2 through 106.7. One has to realize that this is only to be used as a framework to which other describable anomalies could be added, which result from aberrant rather than arrested development.
The most common forms of these anomalies are incomplete partition of the cochlea and relatively mild dysplasias of the lateral SCC.
A dysplastic cochlea and vestibular system will frequently coexist. When the developmental arrest occurs after 7 weeks of gestation, isolated anomalies of the vestibular system can occur (Fig. 106.8). In mild varieties, there may be only a more bulbous lateral SCC and vestibule than usual. Malformations of the superior and posterior SCC also usually involve the lateral SCC, as the lateral SCC is the last to be formed. Malformations consist of narrowing, foreshortening and widening, assimilation with the vestibule, partial or total absence, and dehiscence.
Since the 1990s, a deficient modiolus as seen on CT or MRI and other more subtle internal derangements of the cochlear anatomy started leading to the recognition of anomalous development.3,4 These and other relatively gross inner ear abnormalities have traditionally been detected by visual inspection; thus, even more subtle dysplasias can remain overlooked.
More recently, measurements of the cochlear and vestibular systems have contributed to the detection of cochlear and/or vestibular dysplasia. To increase sensitivity, measurements of the cochlear height on coronal images and of the lateral SCC bony island width should be performed. The height of a normal cochlea ranges from 4.4 to 5.9 mm. Cochlear hypoplasia (<4.4 mm) is associated with SNHL. A dimension of the bony island of the lateral SCC <2.6 mm is consistent with lateral SCC dysplasia. There is no consistent correlation between lateral SCC dysplasia and SNHL (Fig. 104.1). Measurements above the range of normal for these structures may have no practical benefit in identifying inner ear dysplasias.2
Modern imaging is constantly improving. Anatomic abnormalities such as incomplete separation of the scala tympani and vestibuli, subtle modiolar deficiency, and/or an abnormal cochlear nerve aperture relative to the cochlear base may be observed independent of an abnormal measurement such as the cochlear height. Normal measurements do not exclude dysplasia; they are only another tool in a multifactor risk assessment for dysplasia. A complete assessment for inner ear dysplasia should use a limited number of measurements in addition to observations for subtle dysmorphic features such as those just mentioned. With, such a system, anatomic imaging will provide a reasonable but still imperfect means for such screening until better tools such as genetic screening take over or augment anatomic observations at some point.

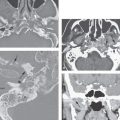
FIGURE 106.3. Cochleovestibular anomaly due to arrest of growth in the fourth gestational week on axial (A,B) and coronal (C,D) CT images. A large cystic structure without internal architecture represents a common cavity for the cochlea and vestibule. In some cases, the semicircular canals are formed.
Related posts:
 Preseptal Compartment: Acute and Chronic Infections and Noninfectious Inflammatory Conditions
Preseptal Compartment: Acute and Chronic Infections and Noninfectious Inflammatory Conditions
 Necrotizing (“Malignant”) Otitis Externa
Necrotizing (“Malignant”) Otitis Externa
 Cervicothoracic Junction: Brachial Plexus Trauma
Cervicothoracic Junction: Brachial Plexus Trauma
 Oropharynx: Introduction
Oropharynx: Introduction
 Branchial Apparatus Developmental Anomalies
Branchial Apparatus Developmental Anomalies
 Oral Cavity and Floor of the Mouth: Malignant Tumors
Oral Cavity and Floor of the Mouth: Malignant Tumors
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


