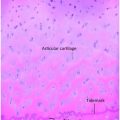
Figure 8.1 Rickets, radiologic–pathologic correlation. Images in 4-year-old child with rickets. A, Frontal radiograph of distal femur shows fraying and cupping of metaphysis, widening of physis, and partial loss of opacity of zone of provisional calcification. B, Corresponding photomicrograph (hematoxylin-eosin [H-E] stain; original magnification, X1) shows accumulation of cartilage cells in physis. Low (C) and medium (D) power photomicrographs (H-E stain; original magnification, X2 and X4, respectively) of sections of physis (boxed regions in B and C, respectively) show tongues of cartilage (*) extending into metaphysis (M). C = chondro-osseous junction, E = epiphysis, M = metaphysis, P = physis. (Images courtesy of Ellen M. Chung, MD, Armed Forces Institute of Pathology, National Museum of Health and Medicine.) (With permission from Perez-Rossello JM, McDonald AG, Rosenberg AE, Tsai A, Kleinman PK. Absence of rickets in infants with fatal abusive head trauma and classic metaphyseal lesions. Radiology. 2015; Epub 2015/02/17.)
The most common cause of osteomalacia in children is vitamin D deficiency rickets but there are several other forms of the disease including hypophosphatemic rickets (vitamin D-resistant rickets) and disorders of vitamin D synthesis and/or action. This chapter focuses on osteomalacia and rickets in the infants and young children where the primary manifestations of the metabolic disturbance (metaphyseal irregularities) and superimposed fractures are relevant to the differential diagnosis of abuse. Rickets is also discussed elsewhere in the context of miscellaneous form of abuse and neglect (see Chapter 23).
Biochemical background: calcium homeostasis
Vitamin D and parathyroid hormone (PTH) are the regulators primarily responsible for calcium homeostasis in humans.
Vitamin D
Vitamin D is synthesized in the skin, and is found in some foods, either naturally (e.g., fish) or through fortification (e.g., in milk) (Fig. 8.2) (6). In the skin 7-dehydrocholesterol is converted by ultraviolet light and heat to vitamin D3 (cholecalciferol). Vitamin D2 (ergocalciferol) is produced by some phytoplankton, invertebrates, yeasts, and mushrooms in response to ultraviolet light irradiation. Both vitamin D2 and D3 are converted to 25-hydroxyvitamin D (25D) by the 25-hydroxylase enzyme in the liver. 25D is inactive and is further converted to the active form of vitamin D, 1,25-dihydroxyvitamin D (1,25D2), by 25(OH)D-1-alpha-hydroxylase in the kidney. 1,25D2 carries out most of its action by binding to the vitamin D receptor (VDR) in cells. The primary action of 1,25D2 is to stimulate active intestinal absorption of calcium and phosphorous in the duodenum when calcium intake is low. When dietary calcium levels are moderate or high, passive, paracellular intestinal calcium transport in the jejunum and ileum is responsible for the bulk of calcium absorption and is not dependent on 1,25D2.
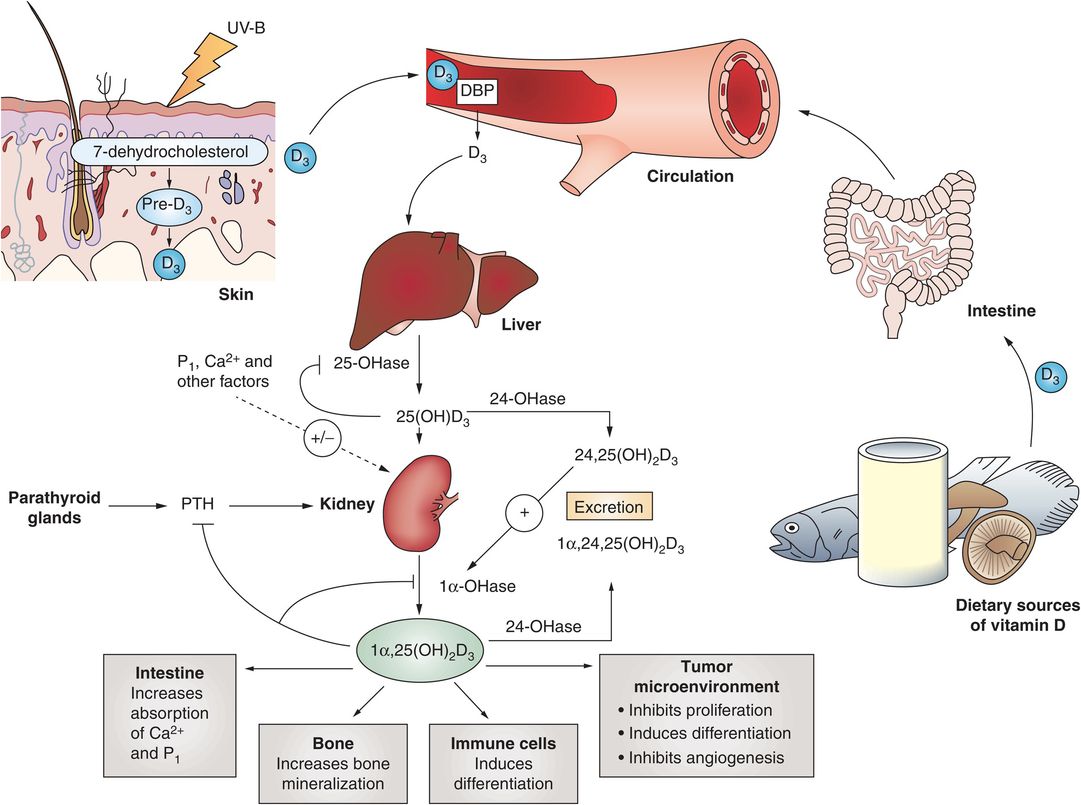
Figure 8.2 Summary of the synthesis, metabolism, and actions of vitamin D. (With permission from Deeb KK, Trump DL, Johnson CS. Vitamin D signaling pathways in cancer: potential for anticancer therapeutics. Nat Rev Cancer. 2007;7(9):684–700.)
The 25D levels reflect vitamin D status, vary with seasons and sun exposure, and are found in concentrations that are 1000-fold greater than that of the active 1,25D2. Vitamin D deficiency is defined by levels of 25D. In a recent statement by the Institutes of Medicine, a 25D level above 20 ng/mL is considered adequate for bone health (7).
Parathyroid hormone
PTH is secreted by the four parathyroid glands, which reside in the neck surrounded by the thyroid gland. Secretion of PTH is regulated by the calcium-sensing receptor (CaSR) in a classic negative feedback-loop manner. The binding of calcium to the parathyroid gland suppresses PTH secretion, whereas low concentrations of calcium in the blood result in decreased binding of calcium to the CaSR, stimulating PTH release. PTH acts on the bone to release calcium. PTH also stimulates reabsorption of calcium in the kidney and stimulates the conversion of 25D to 1,25D2, which in turn increases intestinal absorption of calcium. Together, the actions of PTH lead to increased blood calcium levels, increased binding of calcium to the CaSR, and decreased secretion of PTH from the parathyroid gland.
Calcium homeostasis in the fetus
The fetus accrues 20–30 g of calcium through gestation, most of it (about 80%) in the third trimester (8). This high rate of calcium accrual in the third trimester explains why premature infants, who are born before the third trimester has been completed, have a significant deficit in calcium at birth.
Calcium homeostasis in the fetus differs from the postnatal process in that placental parathyroid-related protein (PTHrP) plays a central role in mineralization of the fetal skeleton, whereas vitamin D is less important (9). This is not surprising since the placenta provides all of the nutrients for the fetus, and there is essentially no intestinal source of calcium in the fetus; amniotic fluid which the fetus swallows has little calcium. The placenta is the primary source of calcium in the fetus, and placental PTHrP stimulates the active transport of large amounts of calcium and phosphorus against a concentration gradient from the mother to the fetus. As a result, calcium and phosphorus levels in the fetus are higher than maternal levels and are maintained at this higher level, even in the presence of maternal hypocalcemia.
The fetus synthesizes PTHrP as well as PTH. Fetal PTHrP plays a role in chondrocyte differentiation and skeletal development. Fetal PTH plays a role in fetal bone mineralization and increases in response to fetal hypocalcemia, similar to the actions of PTH after birth.
Vitamin D and vitamin D receptor (VDR) are not required for fetal skeletal development or mineralization and are not important for fetal calcium and bone homeostasis. 25D crosses the placenta and fetal 25D levels reflect maternal vitamin D status. 1,25D2 does not cross the placenta; it is produced in the placenta and fetal kidneys, and 1,25D2 levels are low in the fetus. Intestinal absorption of calcium in the fetus is trivial since only amniotic fluid is absorbed in the fetal intestines, and what little calcium is absorbed is present is transported passively (non-vitamin D mediated).
The lack of a role for vitamin D in utero is illustrated in animal studies where offspring of vitamin D deficient rats, mice, and pigs have normal fetal blood calcium, phosphorus, PTH, weight, and skeletal mineral, and calcium content (10). VDR-deficient animals have normal calcium homeostasis and bone mineralization, and there is no difference in morphology, physeal architecture, and periosteal thickness compared with controls (10). Children with genetic disorders of vitamin D metabolism – including 1-alpha-hydroxylase deficiency in which there is an inability to convert 25D to 1,25D2 resulting in low 1,25D2 levels, and hereditary 1,25D2 resistant rickets due to mutations in VDR and end-organ resistance to 1,25D2 – are normal at birth and have normal calcium levels despite either lack of active vitamin D or vitamin D action (11–14). Hypocalcemia, hypophosphatemia, and rickets develop later in the first year of life as intestinal calcium transport switches from passive to active, dependent on 1,25D2.
In addition, there is evidence that vitamin D deficiency in the mother does not affect the neonate. The total calcium content of newborns dying of obstetric accidents was the same in neonates born to normal mothers as it was in neonates of mothers who had extreme vitamin D deficiency (15). BMC does not differ at any site in babies within 15 days of life in vitamin D sufficient versus deficient mothers (16). Finally, randomized trials of vitamin D supplementation during pregnancy find no effect on cord blood calcium or phosphorus, anthropometric measurement, or radiologic evidence of rickets. In a randomized controlled trial (RCT) of 1000 IU vitamin D versus no supplement in 126 Asian mothers (17), despite large differences in cord blood 25D levels in the treated group (55 ng/mL) compared to the control group (4 ng/mL), there were no differences in infant serum calcium levels between the groups. In addition, craniotabes was found in both the control group (six infants) and the treated group (two infants), and there was no evidence of rickets in the five infants in the control group with hypocalcemia. Another trial studied vitamin D supplementation during pregnancy (400, 2000, or 4000 IU per day) in 350 women (18). Although neonatal 25D levels were significantly correlated with maternal 25D levels, and differed significantly by treatment group (18.2 ng/mL in the control group, 22.8 ng/mL in the 2000-IU group, and 26.5 ng/mL in the 4000-IU group), there was no difference in clinical status of the newborns.
Calcium homeostasis in the neonate
At birth, calcium levels in the newborn are higher than in the mother (9, 19). After the umbilical cord is cut there is sudden loss of placental PTHrP-mediated calcium transport resulting in a decrease in calcium concentrations within 24–36 hours. The neonate switches to dependency on PTH to mobilize skeletal calcium stores and increase renal calcium reabsorption, and to induce intestinal calcium absorption. This early drop in calcium leads to an increase in PTH to adult ranges by 24–48 hours, increasing calcium levels and 1,25D2 levels, and by 3–7 days calcium levels increase to childhood levels. Within this time period the neonate switches from dependency on placental calcium transport to dependency on intestinal calcium absorption. Initially the intestinal absorption of calcium in the newborn is passive and not dependent on vitamin D. This explains why preterm babies do not respond to treatment with calcitriol (1,25D2). As the neonate matures through the first year of life, passive absorption of calcium declines and there is up-regulation of the VDR and calcium-transporting genes and proteins. By the time of weaning the baby has transitioned from primarily passive calcium transport to a combination of passive and active transport.
Rickets: the basics
The classic features of vitamin D deficient rickets occur as a result of hypocalcemia, which leads to increases in PTH. In order to maintain calcium balance in the face of hypocalcemia due to decreased intestinal absorption of calcium due to vitamin D deficiency, PTH stimulates osteoclastic activity to release calcium from bone. Bone demineralization is characterized by thinning of the cortices, coarse trabeculation, and increased radiolucency of the bones.
The specific radiographic alterations of rickets result from the lack of substrates for mineralization of cartilage matrix and osteoid in the zone of mineralization (calcification) of the physis and the primary spongiosa of the metaphysis. These combined regions comprise the zone of provisional calcification (ZPC). Although some anatomists and pathologists have used the term “ZPC” interchangeably with the zone of mineralization of the physis, we will use the broader classic radiographic definition in this discussion (see Chapter 1) (20–22). In rickets there is loss of the normal opacity of the ZPC. Additionally, the subperiosteal bone collar (SPBC) surrounding the periphery of primary spongiosa and adjacent physeal margin fails to mineralize, resulting in loss of the characteristic metaphyseal “bone bark” described by Laval-Jeantet et al. (23, 24). Extension of noncolumnar tongues of unmineralized cartilage into the primary spongiosa manifests radiographically as an irregular, frayed, or brush-like appearance of the metaphysis (25, 26). The cupped contour of the metaphysis forms from inward bulging and splaying of the metaphysis due to accumulation of hypertrophied cartilage cells and nonmineralized osteoid in the metaphysis (27). Rachitic changes are best seen in the physis of rapidly growing bones such as the wrist, knees, and costochondral junctions (CCJs) (20, 28). Looser zones are rare in infants and young children; in older patients they are seen in longstanding severe osteomalacia and abnormal serum biochemistry is required to differentiate them from insufficiency fractures (29). Long-term effects on the child include delayed bone growth, bowing deformities, and occasionally fractures. The radiographic rachitic changes are generally proportional to the severity of the deficiency, and progressive biochemical changes are related to an altered calcium and phosphorus product (30).
Rickets in the current context
Although there are a variety of disorders of calcium and phosphorus metabolism that result in skeletal alterations in infants and young children (31), this discussion focuses on three forms relevant to the issue of child abuse that can manifest metaphyseal fragmentation and fractures: (1) metabolic bone disease of prematurity; (2) congenital rickets; and (3) nutritional vitamin D deficiency rickets.
Metabolic bone disease of prematurity
The cause of metabolic bone disease in premature infants is multifactorial (32). Premature infants are born prior to the third trimester before most of the calcium accretion in the bone occurs. Additionally, premature infants have difficulty maintaining a normal and balanced calcium and phosphorus ratio. This is usually associated with bronchopulmonary dysplasia (chronic lung disease), prolonged total parenteral nutrition, dexamethasone and diuretic treatment, and cholestatic jaundice (33, 34). Vitamin D deficiency plays a lesser role in metabolic bone disease of prematurity (35). In a study of premature infants with or without rachitic changes and/or fractures, there was no difference in the serum concentration of vitamin D between the groups (36).
The radiographic hallmark of metabolic bone disease of prematurity is demineralization, often referred to as “osteopenia of prematurity” (Fig. 8.3). The overall incidence of demineralization in premature infants is 21–65% (37–39). Infants weighing under 1500 g or born at less than 28 weeks gestational age are at highest risk. Demineralization usually becomes evident at 6–12 weeks of age (40). When the calcium–phosphorus imbalance is severe, rickets can develop. Rachitic changes have been reported in 22–53% of premature infants (36, 38, 39, 41–44). In very small severely premature infants with complicated courses including parenteral hyperalimentation, the ZPC may be preserved and only mild metaphyseal irregularities may be evident, despite profound bony demineralization. The imaging features can also vary depending on the mineral and vitamin constituents of the hyperalimentation solutions, and may be influenced by coexistent copper deficiency (see Chapter 7) (45).
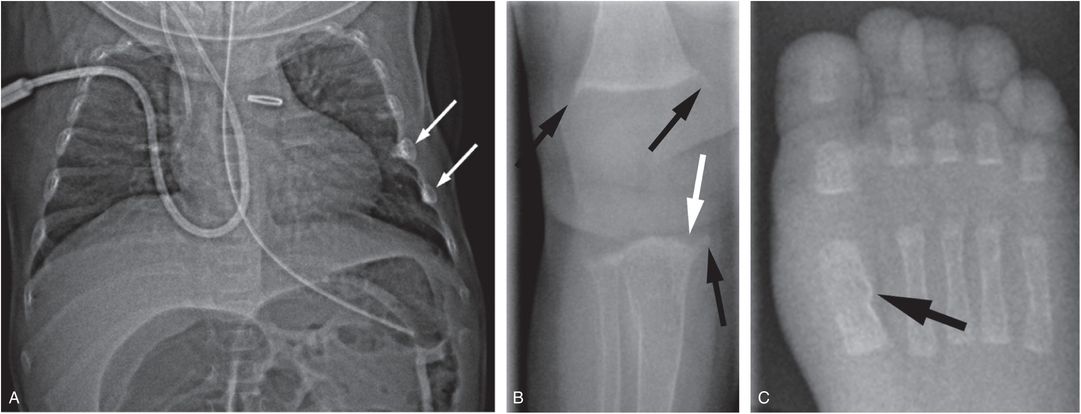
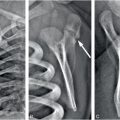
Figure 8.3 Metabolic bone disease of prematurity. Four-month-old former 27-week gestational age, 800-g infant with clinical course complicated by patent ductus arteriosus and necrotizing enterocolitis with bowel perforation. A SS was obtained for evaluation of markedly elevated alkaline phosphatase: 25D = 23 ng/mL (nl: >20); PTH = 573 pg/mL (nl: 10–65); alkaline phosphatase = 1701 U/L (nl: 110–400); calcium = 9.1 mg/dL (nl: 8–10.5); phosphorus 3.1 mg/dL (nl: 3.5–6.6). A, Anteroposteror (AP) view of the thorax shows demineralization and two rib fractures (arrows). B, AP view of the right knee shows metaphyseal irregularity and spurring (black arrows). There is subtle loss of the ZPC along the medial aspect of the proximal tibial metaphysis (white arrow). C, AP view of the right foot shows a healing fracture with SPNBF of the first metatarsal (arrow).
Premature infants with metabolic bone disease are at increased risk of fractures. Early studies of fractures in premature infants found an incidence of fractures, with or without rachitic changes, of 8–22% (38, 41, 43). Improved neonatal care has decreased the incidence of fractures to as low as 1.6% and high as 13% (44, 46). Fractures are reported to be more common in the upper extremity and clinically present with swelling (43, 46). Long bone fractures are usually transverse or greenstick in the metaphysis or diaphysis of the bone. The physis is usually not involved (41). The prevalence of rib fractures in premature infants is low, ranging from 2 to 7% (47–49). Rib fractures are usually found incidentally and typically described in the midposterior, lateral, and anterior arches (46). When multiple rib fractures are encountered, they are often in the same position in multiple ribs, and demineralization is evident (46, 47). Rib fractures in premature infants are rarely related to cardiopulmonary resuscitation (CPR) or chest-tube placement procedures (46, 48).
As discussed elsewhere, prematurity has long been considered a risk factor for abuse (see Chapter 23) (46, 50–52). Fractures occurring on the background of metabolic bone disease of prematurity can present a diagnostic challenge. In some cases, the fracture pattern and distribution are entirely in keeping with the severity of the metabolic alterations, but atypical imaging findings in the context of clinical concerns for abuse should prompt an evaluation by a Child Protection Team (CPT) and a skeletal survey (SS) may be warranted.
Congenital rickets
Congenital rickets is a rare entity seen in infants of mothers with severe osteomalacia, usually encountered in developing countries where women are covered and vitamin D intake is low. In one study of 337.68 million individuals in India (years 1963–2005) 3 newborns were identified with congenital rickets (53). Profound maternal hypocalcemia secondary to prolonged and severe vitamin D deficiency is the major cause of congenital rickets (54). Maternal vitamin D deficiency and hypocalcemia are usually more severe in the mother than in the fetus, since during pregnancy there is preferential calcium transfer to the fetus (55). In the presence of severe maternal hypocalcemia, the transfer of calcium to the fetus may be decreased leading to fetal hypocalcemia and subsequent fetal hyperparathyroidism. Fetal hyperparathyroidism leads to release of calcium and phosphorus from bone. Congenital rickets has been reported in infants of mothers treated with prolonged infusion of magnesium sulfate for tocolysis (56), untreated maternal renal insufficiency (57, 58), severe malnutrition, and vitamin D deficiency (59, 60).
It is important to note that congenital rickets is not due primarily to vitamin D deficiency in the fetus, as the fetus does not absorb calcium from the intestines – rather it obtains calcium through the placenta under regulation by PTHrP. Instead, congenital rickets is due to severe maternal hypocalcemia which leads to fetal hypocalcemia and secondary fetal hyperparathyroidism. The maternal hypocalcemia must be severe enough to cause fetal hypocalcemia, since placental PTHrP-mediated transfer of calcium from the mother to the fetus occurs in the presence of maternal hypocalcemia.
Infants with congenital rickets typically present from birth to two months of age. They have the expected biochemical changes seen in rickets: vitamin D deficiency, elevated PTH, and hypocalcemia. The clinical presentation is usually hypocalcemic seizures or recurrent pneumonias (59, 61, 62). Orbak et al. reported eight mother/infant pairs with osteomalacia/rickets. The infants presented at 1–2 months of age with hypocalcemic seizures. All mothers and infants had vitamin D deficiency (25D <20 ng/mL). All the infants had metaphyseal rachitic changes and biochemical abnormalities. A large survey in India identified 165 mothers with osteomalacia (53, 55). In this group, three infants had neonatal hypocalcemic seizures and three infants had congenital rickets. These infants were born with frontal and parietal bossing, swollen wrists and ankles, and wide anterior fontanels. Radiographs showed rachitic changes of fraying and cupping of the metaphysis. Laboratory studies showed low vitamin D, elevated PTH, and hypocalcemia.
Soliman et al. studied 10 newborns in the first 10 days of life with symptomatic hypocalcemia and vitamin D deficiency (25D levels 13.2 ± 3.8 ng/mL) and maternal vitamin D deficiency (25D levels 8.1 ± 1.5 ng/mL) (63). The radiologic alterations shown were not those of rickets, but rather nonspecific metaphyseal lucent bands with preservation of the ZPC. No fractures were noted. In this study maternal calcium levels were not measured and thus maternal hypocalcemia leading to fetal and neonatal hypocalcemia was not documented.
Reports of fractures in congenital rickets are rare. Rib fractures have been reported in infants of mothers with severe osteomalacia due to renal disease and hypocalcemia (57, 58, 64). A greenstick femur fracture and a radius fracture have also been reported (64, 65). All the fractures reported occurred in the background of metaphyseal rachitic changes.
Nutritional rickets
The most common cause of rickets in children is nutritional rickets, usually due to vitamin D deficiency, but at times due to calcium deficiency, especially in countries such as South Africa, Gambia, Nigeria, and India (66–68). Since maternal vitamin D crosses the placenta freely, infants’ vitamin D levels at birth reflect maternal vitamin D stores (69). Vitamin D deficient rickets is most likely to occur when the mother is vitamin D deficient during pregnancy. Consequently, the infant has low vitamin D stores at birth, is unable to produce endogenous vitamin D due to dark skin pigmentation and/or limited sun exposure, is solely breast fed (breast milk has <50 IU/vitamin D per liter), and the diet is not supplemented with vitamin D (70, 71). Rachitic changes usually present between 6 and 24 months but can be seen as early as 3 months of age, and these young affected infants should not be incorrectly diagnosed as having congenital rickets (67, 72–74).
Vitamin D deficient rickets can be roughly divided into three overlapping stages. In stage 1 decreased 25D leads to decreased 1,25D2 and hypocalcemia, and PTH begins to mobilize calcium from bone. If the demand for calcium is high, as in a period of rapid infant growth, and PTH cannot compensate for the calcium requirement, the infant can present with hypocalcemic seizures. Radiographs are often normal, but may occasionally reveal demineralization and rachitic changes (Fig. 8.4) (31, 75, 76).
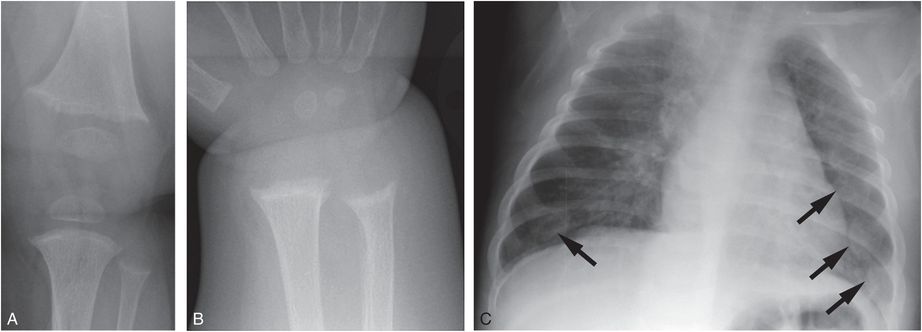
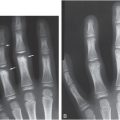
Figure 8.4 Vitamin D deficiency rickets. A seven-month-old infant presenting with a hypocalcemic seizure. The infant was solely breastfeed without vitamin D supplementation. Frontal radiographs of the knee (A) and wrist (B) show loss of the ZPC, fraying, and cupping of the metaphyses. C, AP view of the chest shows flaring of the CCJ (arrows), corresponding to the “rachitic rosary” evident clinically: 25D was < 7 ng/mL; PTH = 356 pg/mL; alkaline phosphatase = 599 U/L; calcium = 4.6 mg/dL.
In stage 2, secondary hyperparathyroidism develops which normalizes the calcium levels, there is PTH-mediated increased conversion of 25D to 1,25D2, and there is hypophosphatemia. Radiographic and clinical manifestations of rickets become more evident at this stage. In stage 3 there is further increase in PTH without effect and the severe manifestations of rickets ensue.
There are limited data defining the 25D concentration at which rickets becomes evident (77). In a small study of nine patients with nutritional rickets the level of 25D correlated with the severity of the rachitic changes. Mild rachitic changes were seen with vitamin D levels between 15 and 21 ng/mL and overt rickets with moderate to severe deficiency between 8 and 14 ng/mL (78). Although calcium homeostasis is the major driver of nutritional rickets, the ratio of calcium and phosphorus has a role in the appearance of radiologic rachitic changes. In a study of 40 infants and young children ages 7–23 months with vitamin D deficiency and normal calcium and phosphate levels, the radiographs showed a normal appearance of the metaphyses and mild widening of the physis (30). With normal calcium but low phosphate, lucency of the metaphysis with mild fraying and cupping occurred. Finally, when both calcium and phosphate were low there was complete loss of the ZPC, metaphyseal cupping, and demineralization. Arnaud et al. noted similar findings (78).
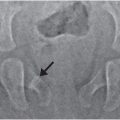
In several recent studies infants with vitamin D deficiency were screened for rachitic changes with hand and knee radiographs. In a Boston study, 44 (12%) of 365 healthy infants and children (8–24 months old) had vitamin D deficiency (25D <20 ng/mL) (71). Rachitic changes were seen in only 2 (5%) of the 40 patients screened (79). The rachitic findings in the two patients were subtle; partial lucency of the ZPC, with or without loss of the smooth metaphyseal margin (Fig. 8.5). None of the cases showed complete loss of the ZPC, metaphyseal fraying, or widening of the physis. Furthermore, there were only two cases in which there was unanimous or majority agreement that there was definite demineralization (79). In a Seattle study, 246 predominantly breast-fed children (6–15 months old) were screened for rickets with alkaline phosphatase levels. Eighteen of the 33 children with elevated levels had radiographs of the wrist and knees. Only four children had rachitic changes; three had loss of the ZPC, with or without metaphyseal cupping and fraying and one had “frank rachitic changes” (80). In a Louisiana study, none of the 80 infants (0–6 months old) who had low vitamin D levels demonstrated rachitic changes on hand radiographs (81). A higher prevalence of rachitic changes was found in a study in India; 48 of 98 full-term healthy infants (2.5–3.5 months old), had severe vitamin D deficiency (25D <10 ng/mL). Wrist radiographs were performed in 33 patients and changes described as metaphyseal widening or a “healing line” were reported in 10 (30%) patients (82). The authors’ criteria for rickets were not in keeping with classic radiographic findings.
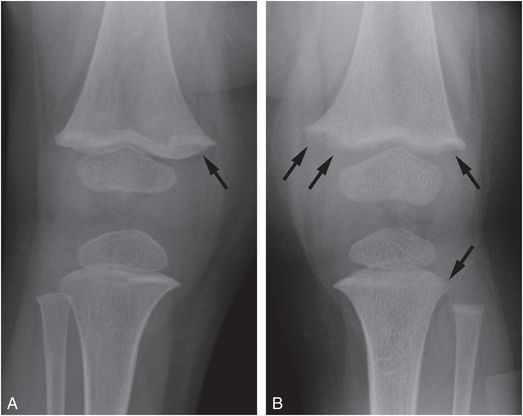
Figure 8.5 Mild rachitic changes in the two vitamin D-deficient infants. A, A 10-month-old infant with a 25D level = 11 ng/mL. AP radiograph of the right knee shows partial lucency of the ZPC (arrow). B, An 11-month-old infant with 25D level = 7 ng/mL. AP view of the left knee shows partial lucency of the ZPC and loss of the metaphyseal margin in the medial and lateral distal femoral metaphysis and the lateral proximal tibia metaphysis (arrows). Note the absence of metaphyseal fragmentation. (With permission from Perez-Rossello JM, Feldman HA, Kleinman PK, Connolly SA, Fair RA, Myers RM, et al. Rachitic changes, demineralization, and fracture risk in healthy infants and toddlers with vitamin D deficiency. Radiology. 2012;262(1):234–41)
While these studies show that vitamin D deficiency is common in the infant population, radiographic findings of rickets are rare and subtle. It is difficult to draw any firm conclusions regarding the prevalence of rickets from the publications due to the varied diagnostic criteria and the limited quality of the radiographs published.
Fractures in vitamin D deficiency rickets are rare (Fig. 8.6).
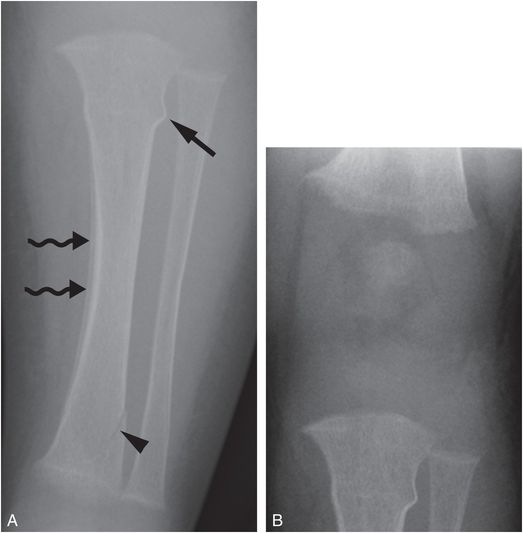
Figure 8.6 Vitamin D deficiency rickets. A four-month-old infant with irritability during diaper changes, transferred for a CPT consultation and further evaluation. A, AP radiograph of the left tibia shows a buckle fracture of the proximal tibial metadiaphysis (straight arrow), an oblique fracture of the distal tibial metaphysis (arrowhead), and diffuse demineralization. Subperiosteal new bone in the medial tibial diaphysis is likely physiologic (wavy arrows). B, AP view of the knee shows mild fraying of the metaphyses and loss of the ZPC, most evident in the femur. The biochemical studies revealed low 25D = 7 ng/mL; elevated PTH = 289 pg/mL; elevated alkaline phosphatase = 1376 U/L; normal calcium = 8.9 mg/dL.
In a large ultrasonography (US) study of fractures in children less than 36 months of age, the causes of the fractures due to metabolic abnormalities, including rickets, was (0.12%) versus falls (50%) or abuse (12%) (83). In a study of 500 infants and children less than 2 years old with clinical findings of rickets, Abanamy and others found only one fracture (84). Chapman et al. studied the prevalence of fractures in 45 children, 2–24 months old, mostly with nutritional rickets. Eighty-three percent of the children did not have fractures, 5% had a single fracture, and 13% had 2–4 fractures (85). The fractures were seen in mobile infants and in the setting of obvious rachitic changes, including metaphyseal fraying and cupping. Perez-Rossello and others found no fractures in their 40 infants and toddlers with vitamin D deficiency (79). Agarwal et al. reviewed wrist, knee, and pelvis radiographs in 25 toddlers (1.0–2.9 years old) with nutritional rickets and found fractures in 2 (8%); each had florid rachitic changes (86).
Rickets versus abuse
Although rickets has long been in the differential diagnosis of child abuse, its profile in this context has been heightened in recent years with the emergence of vitamin D deficiency as a major public health issue (87–89). The high prevalence of a medical disorder that has the potential to affect bone health has coincided with the growth of an active community of defense attorneys and hired medical experts that may serve to provide alternative explanations for “unexplained” fractures in infants.
In 2008, the Keller and Barnes commentary “Rickets vs. abuse: a national and international epidemic” drew considerable attention because it asserted that vitamin D deficiency and insufficiency regularly resulted in congenital rickets – causing multiple fractures resembling those classically associated with infant abuse (90–92). The publication was accompanied by a strongly critical editorial stating “that the connection made by Keller and Barnes between ‘rickets’ and fractures they consider to be similar in appearance to those seen in child abuse is not based on any scientific data” (93). Furthermore they commented that “Unfortunately, the current scenario is reminiscent of Paterson’s ‘temporary brittle bone disease’ ” (see Chapter 13). Feldman, who had personal knowledge of three of the four cases described by Keller and Barnes, strongly criticized the commentary in a subsequent letter to the editor, based in part upon the authors’ failure to disclose that “Neither Dr. Keller nor Dr. Barnes provided clinical care to these children” and that “All the cases came to them as defense witnesses in the legal system” (94). Keller and Barnes offered a response to Feldman and to the commentaries that accompanied their original publication, reasserting their previously stated views (95–97).
Paterson revised his original views on “temporary brittle bone disease” (“TBBD”) including vitamin D deficiency as a possible underlying factor (98). This publication also stimulated an accompanying commentary (99) and numerous letters to the editor (91, 100–104). Drawing heavily on cases referred for consultation by defense attorneys, Paterson and others have continued to support the view that vitamin D deficiency is a causative factor of “TBBD” and is regularly confused with infant abuse (105–109).
A similar position has been taken by Ayoub et al. in their assertion that the radiologic–histopathologic features of the classic metaphyseal lesion (CML) described by Kleinman et al. in a series of abused infants are in fact not traumatic injuries, but rather manifestations of rickets (110). They offered no original evidence in support of this view. In an accompanying editorial commentary, the AJR stated that Ayoub and associates’ view contradicts the published investigations of Caffey and Silverman that indicate that the metaphyseal lesions are traumatic and due to child maltreatment. The commentary indicates that “Dr. Ayoub and coauthors do not justify their stated conclusion that ‘CMLs’ are not true fractures, but, rather a combination of tissue processing artifacts and misinterpreted findings of healing rickets” (111). Subsequent letters to the editor from a variety of authorities harshly criticized the Ayoub et al. publication (112, 113). Ayoub et al.’s hypothesis was examined in a subsequent study by Perez-Rossello et al. published in 2015 (114). The authors performed an archival review (1984–2012) of the radiologic and histopathologic findings in 46 consecutive infant fatalities referred from the state Medical Examiner’s Offices for the evaluation of possible child abuse. Thirty infants with distal femoral histologic material were identified. Additional inclusion criteria were: (1) the medical examiner determined that the infant had sustained a head injury and that the manner of death was a homicide; (2) at least one CML was evident on SS; (3) CMLs were confirmed at autopsy; and (4) non-CML fractures were also present. Nine infants (mean age: 3.4 months; range: 1–8 months) were identified. Two pediatric radiologists independently reviewed the SSs for rachitic changes at the wrists and knees. A bone and soft tissue pathologist reviewed the distal femoral histologic sections for rickets. The authors found no radiographic or pathologic features of rickets in the cohort. The findings provided no support for the notion of Ayoub et al. that the CML is due to rickets. Rather, they strengthened a robust literature that states that the CML is a traumatic injury commonly encountered in physically abused infants.
An extensive review of the literature found no evidence that vitamin D deficiency can cause multiple fractures (115). In children less than two years of age, Schilling et al. found no association between vitamin D deficiency and the diagnosis of abuse and presence of multiple fractures, including metaphyseal and rib fractures (116). As noted previously, when vitamin D deficiency rickets is present in otherwise normal infants, fractures are uncommon. In a study by Chapman et al., any metaphyseal fractures in children with rickets occurred more towards the diaphysis with an appearance suggestive of collapse from axial load. The fractures did not resemble CMLs, and, importantly, they occurred in the presence of characteristic rachitic fraying and cupping of the metaphyses (85).
In an extensive review of rickets, Shore and Chesney stress that rickets is ultimately a histopathologic diagnosis even though the diagnosis is usually made on clinical, laboratory, and radiologic grounds (73, 74). The radiologic and histologic metaphyseal changes seen in vitamin D deficiency rickets differ from those classically described in abuse (117). Jaffe believed that fractures in the metaphysis are very uncommon in vitamin D deficiency rickets because the higher plasticity of the metaphysis caused by the abnormal increase in unmineralized osteoid is protective, allowing the metaphysis to bend rather than break (20).
A healing CML can have focal extensions of hypertrophic cartilage into the metaphysis (118). These observations contrast with classically described radiologic–pathologic features of rickets, which include the loss of the SPBC (23). A study of 49 infants and young children with rickets found loss of the SPBC in 48 (98%) patients (24). Oestreich reviewed the radiologic appearance of the alleged cases of vitamin D deficiency rickets in the commentary of Keller and Barnes (90) and found that none of the cases had loss of the bone collar (119).
Five years after the original Pediatric Radiology commentary “Rickets vs. abuse: a national and international epidemic,” the editors concluded; “Looking broadly beyond Keller and Barnes, since 2008 no study in the literature has shown that fractures considered to be highly specific for child abuse are caused by vitamin D insufficiency or deficiency …. The hypotheses set forth in ‘Keller & Barnes’ thus remain hypothetical and unsubstantiated” (90, 120).
Bone mineral density and fracture risk
Metabolic bone disease, either from decreased osteoid matrix or osteomalacia, causes demineralization, which may increase the risk for fracture. Quantitative measurements of bone mineral density (BMD) correlate with biomechanical properties of bone. Decreased BMD is associated with an increased risk of fracture in adult osteoporosis. The relationship between BMD and fracture risk in children has not been adequately characterized (121, 122). Dual energy x-ray absorptiometry (DXA), quantitative US, and quantitative CT (QCT) are used to measure bone density in children (123). However, as previously noted, the diagnosis of osteoporosis in children requires a history of significant fractures as well as a low BMD (5, 124, 125).
Rigorous bone densitometry research studies in infants and young children are scarce. Overall, premature infants (126–128) and chronically ill children (129) have decreased BMD and increased risk of fractures. Treatments with anticonvulsants, steroids, or chemotherapy are also risk factors. A handful of studies in infants and children with rickets and vitamin D deficiency have shown no difference between their BMD and that of controls (130–132).
Most densitometry research has been performed with DXA, which has limitations in children. DXA gives an areal two-dimensional (2D) BMD and is, thus, dependent on patient size. Since child size is not only age-dependent but also related to pubertal status, even age-specific norms may not be entirely accurate. DXA frequently underestimates BMD in children; therefore, a low BMD may be over-diagnosed in the pediatric population (133, 134). QCT is a more accurate and efficient technique for the assessment of bone density and is well suited to a pediatric population (135). First, QCT achieves a true three-dimensional (3D) volumetric density calculation (136–138). Second, it can differentiate and calculate the density of cortical and trabecular bone, while DXA does not distinguish between the two (139). Third, it furnishes critical structural and functional information including the biomechanical properties of bone (i.e., tension, bending, and torsion), which correlate with QCT density in children (140). Fourth, QCT is able to provide bone geometry quantification from overall dimensions of bones to trabecular microarchitecture or cortical bone porosity (141, 142). Finally, studies have shown that QCT is ideally suited for evaluating the dynamic composition, structure and function of the growing skeleton (143).
In the past, utilization of QCT in pediatrics was limited because it delivered a higher radiation dose than DXA (144, 145). Advances in CT technologies permit performance of the studies with significantly decreased radiation dose without compromising diagnostic accuracy or image quality (146, 147). High-resolution QCT techniques in peripheral sites such as the tibia can be achieved with doses lower than with DXA (148, 149). Future QCT studies in normal infants and toddlers, as well as those patients with metabolic bone disease should provide accurate and precise data that can assist in estimation of fracture risk in young children. These studies may be of particular value when fractures are identified in cases where child abuse must be distinguished from metabolic bone disease.
Reflections and conclusions
Despite the high prevalence of vitamin D deficiency/insufficiency in infants and toddlers, this state is rarely associated radiographically with rickets. It should be acknowledged that metabolic bone disease due to vitamin D deficiency may exist in the face of normal radiographs, and further study regarding the consequences of low vitamin D on general bone health is needed. However, in the absence of radiologic features of rickets, multiple fractures in otherwise normal infants, particularly those with the high-specificity patterns detailed elsewhere in this text, should be viewed as strong indications of child abuse (Fig. 8.7).
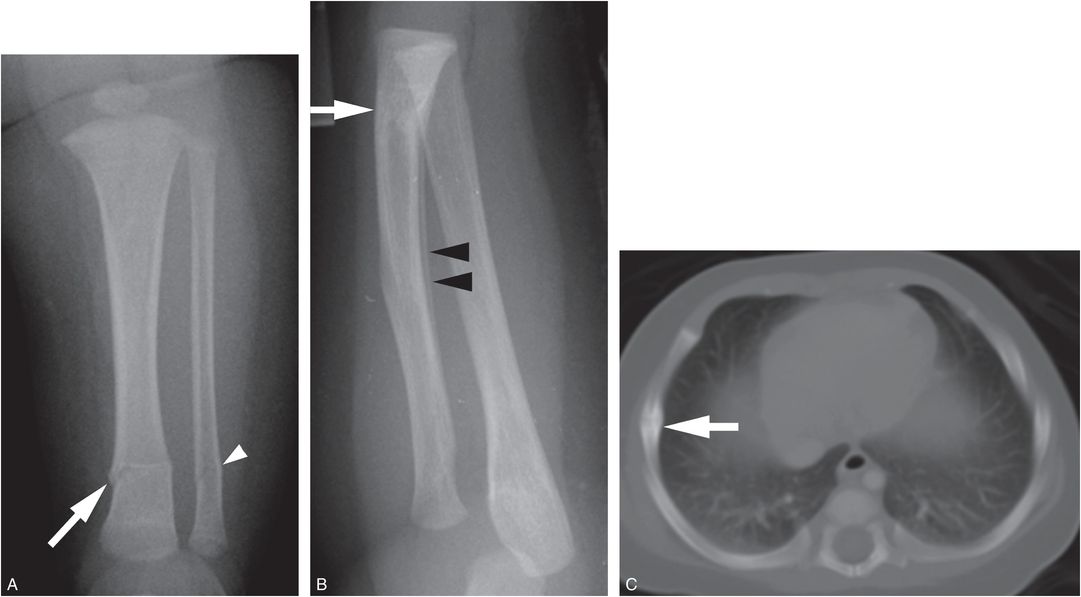
Figure 8.7 Abused infant with vitamin D deficiency. A three-month-old infant not moving the left leg. A, AP view of the tibia shows transverse fractures of the distal tibial (white arrow) and fibular (white arrowhead) diaphyses. B, Lateral view of the forearm from the SS shows a healing transverse fracture of the distal radius with linear medullary sclerosis (white arrow) and SPNBF (black arrowheads). C, CT scan shows a right lateral rib arc fracture healing with callus formation (white arrow). Laboratory values: low vitamin 25D = 5 ng/mL; PTH = 26.6 pg/mL; alkaline phosphatase = 305 units/L; calcium = 11 mg/dL. There are no rachitic changes. The father admitted to causing the fractures.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


