| Type I |
Milder phenotype Normal or close to normal stature Hearing loss in ~50% Demineralization Wormian bones Group 1A: blue sclerae normal teeth Group 1B: dentinogenesis imperfecta (DI) |
| Type II* Lethal in the perinatal period |
| Type III* |
Moderately severe phenotype early Short stature at birth with bowed legs Blue gray sclerae at birth converting to normal white DI common Early severe generalized demineralization Many fractures early and throughout life Progressive spine deformation – codfish vertebrae Skull deformation, poor ossification, Wormian bones Slightly short, thin long bones with thin cortices Extremity bowing/angular deformity “Popcorn” metaphyseal calcifications Exuberant callus formation |
| Type IV* |
Similar to type I Significant short stature early (even in the newborn) Distinctive craniofacial configuration (triangular facies with bitemporal bulging) Fracture onset often prenatal Normal sclerae Hearing loss uncommon Group IVA normal teeth Group IVB DI |
| Type V* |
Normal sclerae Normal teeth Demineralization Moderate to severe bone fragility Moderately deforming Multiple fractures Decreased BMD Wormian bones Wedge-shaped and flattened vertebrae Limited pronation/supination of forearm Anterior dislocation of radial head Calcified interosseous membrane of forearms Hyperplastic callus formation |
| Recessive forms of OI* |
| Usually very severe and most similar to perinatal lethal/severe progressive deforming OI |
* These types are usually obvious and do not pose a problem in the diagnosis of inflicted trauma.
Type I has a mild phenotype usually with normal stature in early life and normal facies. Most cases have blue or gray sclerae, with a developmental hearing loss in about 50% of patients. Dentinogenesis imperfecta (DI) is less common and difficult to diagnose in infants without teeth eruption. Although the first fracture often occurs in the preschool period, radiographically discernible demineralization is always present (Figs. 9.1, 9.2). Ultimately, bowing of long bones often occurs. Most children have 5–20 fractures by the time they reach adolescence. Mild kyphoscoliosis can develop in adolescence in about 20% of cases. Significant Wormian bones are usually present, especially in early childhood.
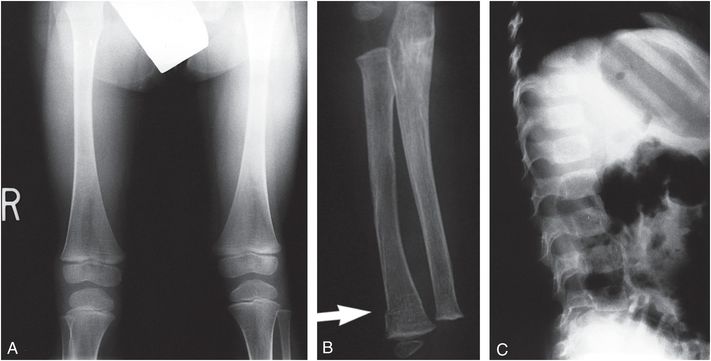
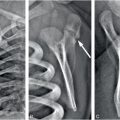
Figure 9.1 Clinically confirmed OI type I, type I collagen analysis negative. No genetic defect discovered at time of diagnosis. A, AP view of the lower extremities in a three-year-old child with generalized demineralization, no fractures, and with relatively normal cortical thickness. B, AP view of the forearm shows demineralization of the radius and ulna with mild cortical thinning. There is a transverse fracture of the distal radius (arrow). C, At the same age, the lateral spine radiograph shows more obvious demineralization.
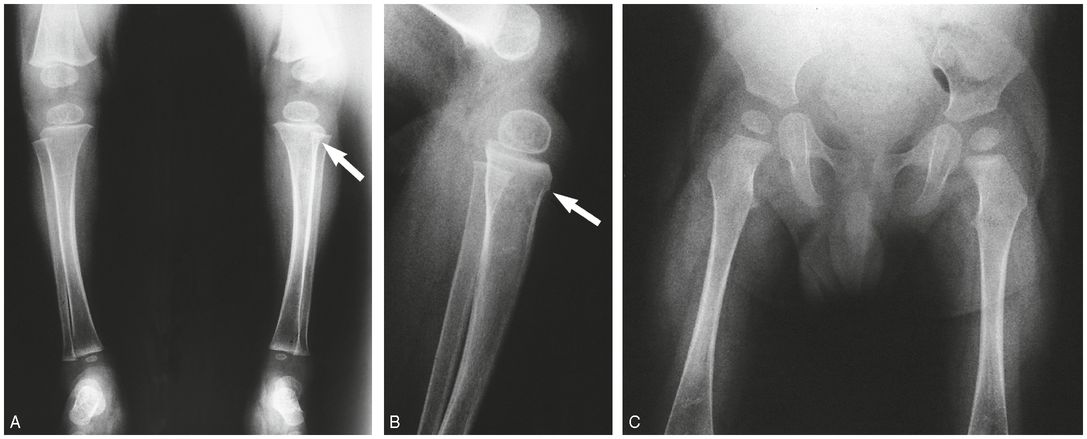
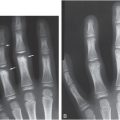
Figure 9.2 Clinically confirmed OI type I, type I collagen analysis negative. No genetic defect discovered at time of diagnosis. A, B, AP and lateral radiographs at seven months of age show a metadiaphyseal insufficiency fracture (arrows) of the left tibia with generalized demineralization and cortical thinning. C, AP radiograph of the pelvis and femurs at nine months of age shows generalized demineralization.
OI type IV has a phenotype similar to type I except for several distinctive features. The craniofacial relationships are often abnormal, with a triangular face, biparietal widening, and a broad forehead. This form of OI is also associated with bluish or grayish sclerae. Radiographically, there may be evidence of platybasia with an “occipital overhang.” Short stature is commonly present (40%) even in the newborn period. The fracture onset can be prenatal, and again, demineralization is generally appreciable on high-quality radiographs (Figs. 9.3–9.7). At times, distinction from type I is difficult, especially in early childhood, and some authors feel that these type IV cases actually belong to the first type. OI type IV is rare, constituting less than 5% of all cases of OI. Taitz estimated the incidence of OI type IV without features of the disease or a family history in infants presenting under 1 year of age to be between 1 in 1 million and 1 in 3 million births (20), but it is unclear whether this figure is still applicable in the era of genetic testing.
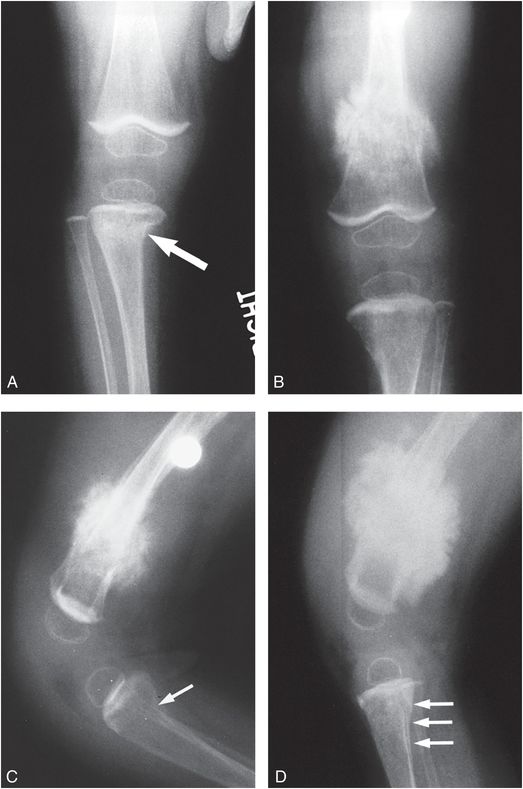
Figure 9.3 Clinically confirmed OI type IV, type I collagen analysis negative. No identifiable defect discovered at the time of diagnosis. A, AP radiograph of the right knee at age 10 months shows generalized demineralization, thin cortices and an impacted transverse metadiaphyseal fracture of the proximal tibia (arrow). AP (B) and lateral (C) views of the left knee show hyperplastic callus at a distal femoral fracture site. There is angular deformity of the proximal tibial metaphysis (arrow). There is obvious generalized demineralization. D, Five weeks later the productive callus around the left femoral fracture has enlarged, producing a pseudotumor. There is SPNBF around a transverse fracture of the proximal tibial metaphysis.
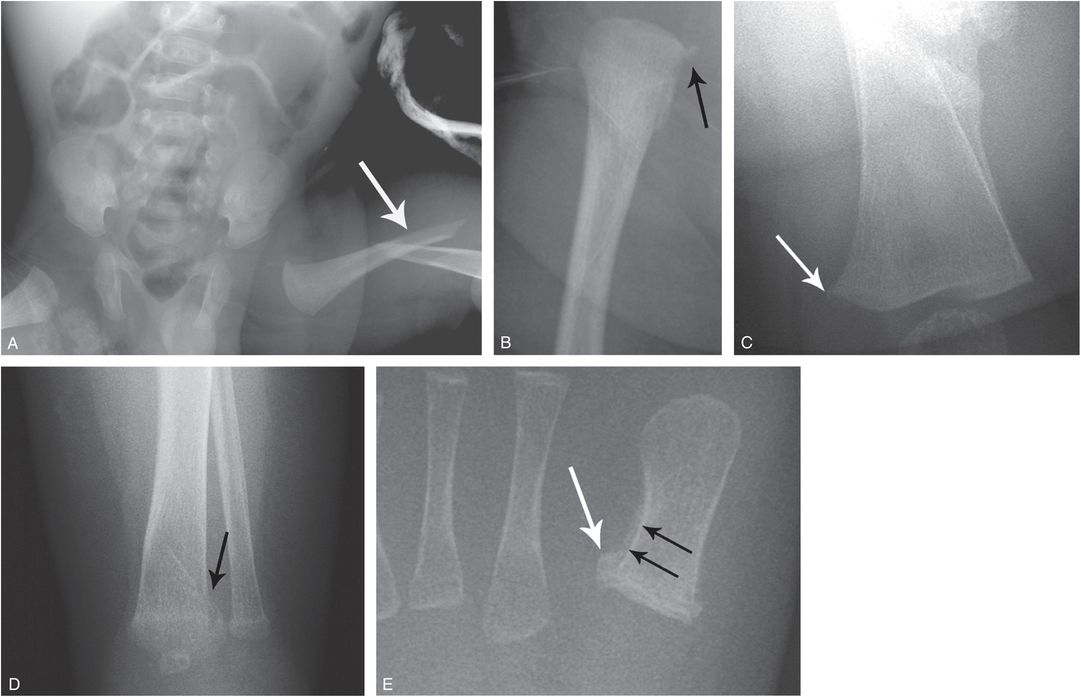
Figure 9.4 OI type IV presenting with suspected abuse. A five-week-old male with one day of swollen left thigh, screaming during diaper changes, and no history of trauma. A left femoral fracture was identified at outside facility and the patient was transported. A, AP view of the pelvis and femur from the SS shows a displaced left femoral shaft fracture (arrow). B, AP view of the right femur demonstrates an osseous fragment (arrow) arising from the right proximal femoral metaphysis. The mineralization appears borderline normal. The sclerae were white and there was no family history of OI. A genetics consult was obtained and collagen testing was advised. A mandated report was filed and the child was placed in foster care. C, One month later, an AP view of the left femur shows robust callus about the fracture and a small osseous fragment arising from the medial aspect of the distal femoral metaphysis (arrow). D, AP view of the left distal tibia shows a nondisplaced acute distal tibial metaphyseal fracture (arrow). E, AP view of the left foot shows a healing metaphyseal fracture of the base of the first metatarsal (white arrow) with SPNBF (black arrows). Demineralization became more evident, raising further concerns of OI. Collagen analysis revealed an abnormality in type I collagen consistent with a skipping mutation of the COL1A2 gene and OI type IV.
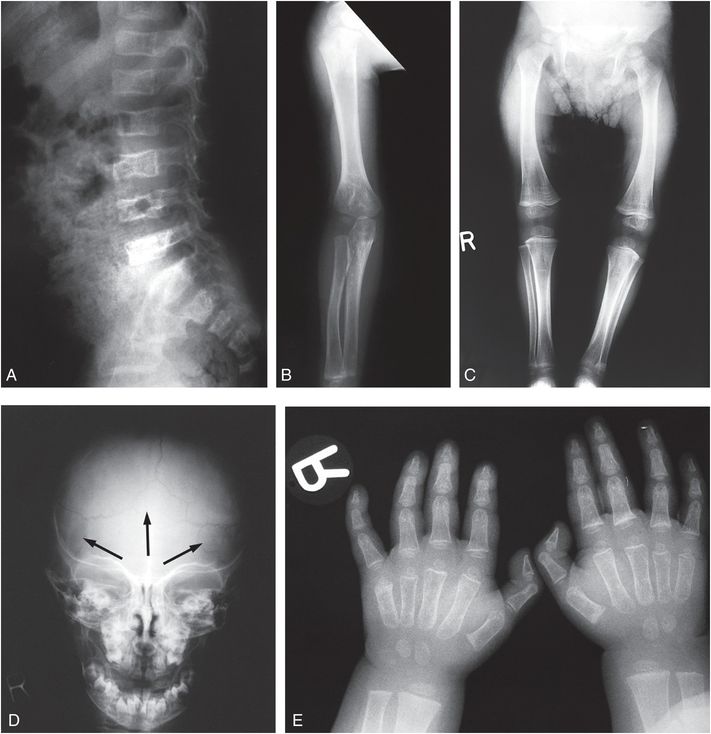
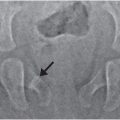
Figure 9.5 Clinically confirmed OI type IV, type I collagen analysis negative. No identifiable defect discovered at the time of diagnosis. A, Lateral radiograph of the lumbar spine in a 14-month-old child shows demineralization with mild vertebral wedging and flattening. B, C, AP radiographs of the right arm and both lower extremities show diffuse demineralization; the cortices are not significantly thin. There is mild rhizomelia and mesomelia, which was supported by clinical findings. D, AP view of the skull radiograph shows multiple large and unusually shaped Wormian bones (arrows) without gross demineralization. E, PA radiograph of both hands also shows generalized demineralization with a normal bone age.
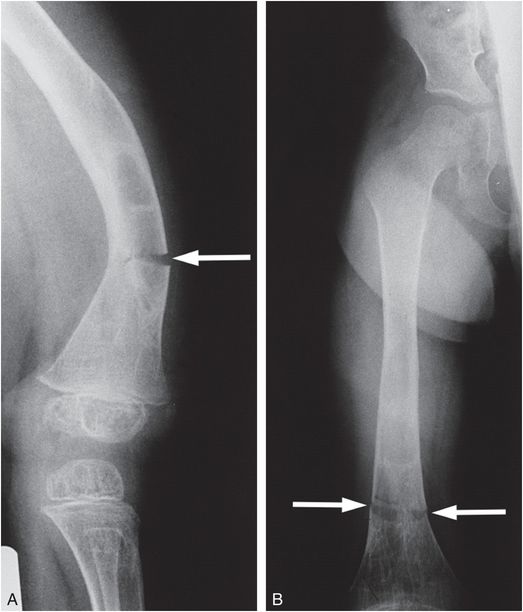
Figure 9.6 Clinically confirmed OI type IV, type I collagen analysis negative. A molecular defect was found. Lateral (A) and AP (B) views of the femur with a transverse insufficiency fracture (arrows) in a demineralized bone through an area of previous fracturing, bowing, and remodeling best seen in B. The bones of the pelvis also show considerable demineralization.
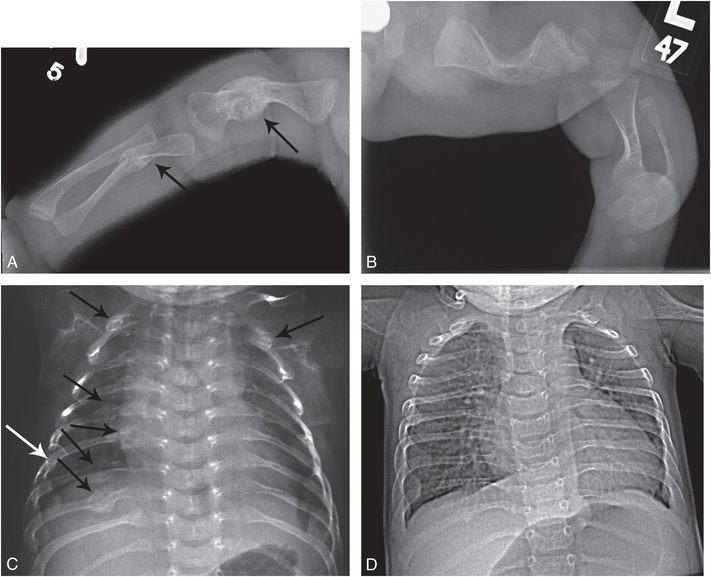
Figure 9.7 OI (type III or severe type IV) with rib and long bone fractures. A one-month-old infant noted to have macrocephaly and long bone bowing on prenatal sonography. Patient was born by spontaneous vaginal delivery and weighed 2 lbs. The arms and legs were short and there were multiple acute upper extremity fractures. The sclerae had a light bluish tinge. Molecular testing revealed a pathogenic heterozygous missense mutation in the COL1A2 gene (c.1378G>A, p.Gly460Ser). There was no family history of OI. A, Frontal radiograph of the right upper extremity reveals mature healing fractures of the shortened humeral and ulnar shafts (arrows). B, AP view of the left lower extremity shows short bowed long bones. C, AP view of the chest demonstrates multiple mature rib fractures involving the lateral and posterior arcs (arrows). Widening of the rib necks at other sites suggests additional old healed fractures. D, AP view of the chest at 10 months of age reveals minimal residual rib deformities. Although the clinical assessment could not differentiate type III from severe type IV, the radiographic features are more in keeping with type III.
Specific discussion of type II and III is not warranted here, as type II is invariably lethal, and type III is severe enough and sufficiently distinct clinically and radiographically not to be considered a mimicker of inflicted trauma (Fig. 9.7). The more recently described recessive forms of OI are also quite severe and are usually prenatal in onset (21). OI type V is an autosomal dominant form of OI that is a rare and moderately deforming type; patients exhibit significant fragility of long bones and vertebral bodies (Table 9.1) (22, 23). These patients have fractures in infancy and have ongoing fractures throughout life. While it would probably be considered too severe to confuse with abuse, it is included here for completeness.
General clinical findings
It is important for radiologists as well as clinicians to understand the major clinical findings in OI. These include the following: blue sclerae, DI, abnormal skin texture, joint laxity, hearing loss, and vessel fragility. The clinical diagnostic criteria may be difficult to ascertain by a nonexpert, just as demineralization can be so easily misinterpreted by a nonradiologist. Clinical geneticists are usually the individuals who have the most experience in not only eliciting the legitimate genetic information about the case, but also defining whether any of the major clinical findings are present. Genetic centers deal with large numbers of OI patients, especially the milder cases (types I and IV) for consultation, as well as other nonorthopedic care. The OI Foundation (http://www.oif.org) also serves as a national referral resource directing patients and physicians to centers with expertise in OI.
Blue, sometimes gray, sclerae are an important sign in many forms of OI (24). They can be a normal finding in early infancy, when the sclerae are normally quite thin. There is also a marked variability in normal scleral color at all ages. Therefore, the diagnosis of true blue sclerae may depend on an experienced clinician, with possible follow-up evaluation necessary (25).
The abnormal skin texture of OI is milder and quite different from the skin of patients with Ehlers–Danlos syndrome or cutis laxa, which represent other connective tissue disorders. The skin has been described as very smooth, soft, sometimes thin, and not really loose, “velvet-like” in texture (26, 27). Similar to Ehlers–Danlos syndrome, there can be joint laxity, including the phalanges. Gentle tugging on the phalangeal tips is associated with movement of the distal, middle, and proximal phalangeal joints, or “telescoping” fingers.
Abnormal hearing is frequently seen in OI and is conductive as well as sensorineural. There may be high-frequency range hearing loss, which some authors believe may be specific for OI. The deafness has its onset in the teenage years and increases in severity with age. Thus, hearing loss is useful only for family history documentation in the very common autosomal dominant forms (28).
DI may be present in all types of autosomal dominant OI (except type IV) and this includes the milder form of OI, type I (29). DI is not seen in the rare recessive forms of OI. Although the radiographic diagnosis of DI can be appreciated by experts, most pediatric radiologists find it difficult to make this diagnosis radiographically by the presence of unerupted primary teeth. In one author’s (R. L.) experience, radiographic DI is not difficult to diagnose before eruption in OI types II and III, but is harder to ascertain in type I. Even after eruption, it is not uncommon to fail to recognize DI on radiographs, only to find that clinically it is present. Therefore, the combination of both clinical and radiographic evaluation is important in the first few years of life. The subgrouping of types I and IV into DI and non-DI groups is not important in determining clinical severity, but often consistently segregates in families with dominant OI. Finally, histologically proved DI without other clinical findings has been reported. DI can manifest as an isolated finding and inherited autosomal dominant disorder and, therefore, by itself does not warrant a diagnosis of OI (30). Other conditions may be associated with DI and include the calvarial “doughnut lesions” syndrome with demineralization and fractures (31); another association of cortical defects, Wormian bones, and DI (32); and spondylometaphyseal dysplasia with DI (odontochondrodystrophy) (33). Joints have been reported to be excessively mobile in some cases of OI. However, the joint laxity is not as severe as that seen in joint hypermobility syndromes such as Ehlers–Danlos syndrome, some of which may be associated with increased fracture risk (34, 35).
Other disorders with brittle bones
There are many other disorders that present with brittle bones either due to under or over mineralization of bone. While OI is very uncommon in the general population, these other brittle bone disorders are extremely rare and usually present in the neonatal period. Also relative to OI types I and IV, these disorders tend to be more severe. Examples include Bruck syndrome, an autosomal recessive disorder characterized by OI and congenital joint contractures (36, 37), and infantile/childhood hypophosphatasia, characterized by bony demineralization, metaphyseal irregularities, and loss of teeth (36, 38–40). Other syndromes, metabolic disorders, and skeletal dysplasias associated with fractures are detailed elsewhere (see Chapters 7 and 8) (34).
Genetics
Despite the explosion in our understanding of the rare severe recessively inherited forms of OI, some cases have unresolved genetics. However, more than 90% of all OI cases, particularly the milder forms (OI types I and IV) are autosomal dominant disorders due to mutations in type I collagen (41). Because of the possibility of dominant inheritance, family history may be important. However, some basic genetic information is necessary to interpret the findings correctly. Although there may be a history of an affected parent with multiple fractures, actual clinical evaluation of the parent as well as radiographic evaluation is important to confirm the diagnosis. Possibly affected siblings should also undergo the same confirmatory evaluation. Since intrafamilial variability can be significant, these avenues must be pursued to establish a correct diagnosis. The vast majority of OI types I and IV cases result from de novo mutations, and therefore parents and siblings are not clinically affected. Also, when more than one sibling is affected and both parents appear normal, one may still be dealing with autosomal dominant OI types I and IV rather than a recessive disorder, because of the possibility of germline mosaicism which occurs in approximately 1.3% of cases of OI (type II) after the birth of one affected child (42). However, the exact incidence of germline mosaicism can be hard to quantify without molecular diagnosis for both autosomal dominant and recessive forms of OI, but should be considered when investigating a family with multiple affected children and unaffected parents.
Chondro-osseous pathology
Bone and cartilage histologic appearances in cases of OI are also not type specific. A broad range of pathologic findings is encountered, including decreased collagen fibril diameter and smaller apatite crystal size on electron microscopy. Conventional histologic studies show changes, including decreased orbicular bone volume, thin cortices, a reduced calcification rate, and thickened periosteum with a defective microvascular system (43). In the International Skeletal Dysplasia Registry’s large experience with chondro-osseous specimens from patients with OI, the region of the physis is never entirely normal. The molecular abnormalities in all types of OI (especially in type III) appear to directly influence not only matrix synthesis but also its organization in bone tissue. The chondro-osseous structure may lead to another diagnosis in cases in which the collagen testing is normal and the patient is still thought to have OI.
Biochemical and molecular aspects
The biochemistry of OI indicates that it is a large group (or family) of disorders. At the present time there are numerous genes that when mutated and inherited in a dominant or recessive manner can produce OI. However, more than 90% of cases result from mutations in the two genes that encode for type I collagen, COL1A1 and COL1A2 (35). The collagens are the predominant proteins of the extracellular matrix of most tissues. To date, there are 29 different known human collagens. Type I collagen is the major collagen of skin, bone, tendon, and blood vessel walls. The distribution of type I collagen correlates with the tissue sites responsible for the signs and symptoms of OI. The long triple helix of the collagen molecule is flanked on each end by a globular amino terminal and a carboxy terminal that function in both the assembly of procollagen and its secretion. These peptides are then removed by proteolytic cleavage in the extracellular compartment (Fig. 9.8). The individual molecules must be normally formed so that they can assemble into an ordered fibril. A mutation that interferes with: (1) expression of the collagen gene; (2) formation of the triple helix; or (3) procollagen secretion can have dramatic effects on the structure and function of the fibrils created, and almost always results in a clinical disorder. The evolutionary relationship of the collagen gene and its protein products provided a unique opportunity to compare mechanisms of mutation in a family of genes and observe the effects of such mutations at the biochemical, histologic, and phenotypic levels.
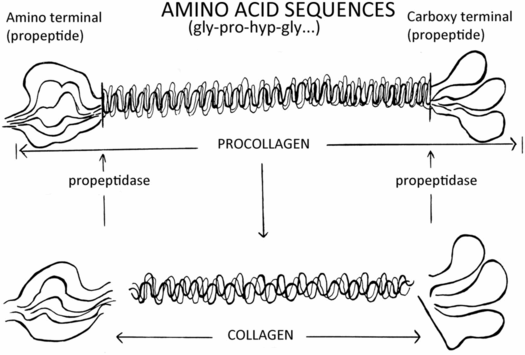
Figure 9.8 Procollagen and collagen molecule processing.
OI is a group of disorders that affects the production, structure, modifications, secretion, and stabilization of type I collagen. Most of the mutations that produce OI result from mutations in the genes that encode type I collagen, COL1A1 and COL1A2. Among type I collagen mutations, two types of defects have been found: (1) quantitative defects characterized by decreased production of normal type I collagen; and (2) qualitative defects in which there are populations of both abnormal type I molecules and structurally normal molecules. The quantitative defects appear to cause the milder forms of the disease and are found primarily in OI type I. On the other hand, qualitative defects have been discovered in OI types II, III, and IV (perinatal lethal, severe, and moderate, respectively). Most of these defects are point mutations that result in substitutions for a glycine residue (41, 44–47) in the triple helix of one of the type I collagen chains. This disruption of the normal amino acid sequence slows the assembly of the molecule, creating a less stable abnormal structure. Consequently, decreased secretion and/or intracellular degradation of procollagen will occur. Quantitative defects that limit the amount of type I collagen may result in decreased deposition of normal collagen in the extracellular matrix and a decreased mineralized extracellular matrix. Qualitative defects, in which abnormal collagen trimers are incorporated into the matrix, usually severely disrupt mineralization. The phenotypic effects of having too little collagen in bone appear to be far milder (OI types I and IV) than those resulting from the presence of abnormal chains of collagen.
OI results from alterations in the genes that encode type I collagen. The two genes are COL1A1 on chromosome 17 and the COL1A2 gene on chromosome 7. Both genes are similar in structure and encode for the approximately 1000 amino-acid repeat (glycine–X position amino acid–Y position amino acid), which is the triple helical domain of type I collagen. Because of the complexity of collagen synthesis and the importance of maintaining the structural motif of the glycine–X–Y repeat, gene alterations that cause a structural change or lead to decreased production of type I collagen result in OI. Hundreds of different mutations have been demonstrated in OI and most individuals (or families) have unique or private mutations. The location of the mutation in the gene, the chain in which it occurs, and the type of amino acid substituted all contribute to the phenotypic spectrum. By far the most common event leading to OI is a point mutation (substitution), resulting in substitution for glycine in the triple helix. Glycine substitutions are seen in OI types II, III, and IV, and, rarely, in type I. In OI types II and III (lethal and severe) the mutations tend to cluster near the carboxy terminus, and the substituting amino acids (e.g., valine and aspartic acid) have a more deleterious effect on helical formation.
Most commonly in OI type I, a glycine amino acid is substituted by a nonsense mutation, or, a stop codon, that leads to decreased production of type I collagen (“a null allele”) (48). Also mutations in the carboxy terminal propeptide region, where the chains associate, prevent incorporation of the abnormal chain and can lead to varying OI phenotypes (49). Patients with OI type IV, the other category important in terms of child abuse, have substitutions for glycine residues, frequently by arginine, cysteine, or serine. Unfortunately, these mutations do not cluster in any specific region. Initially, it was thought that the phenotypes of OI could be attributed to the region of the molecule in which the mutations occurred. However, the phenotype is not solely dependent on location of the mutation but also depends on the chain in which it occurs and the nature of the substituting residue.
It was also once thought that the collagen biochemistry could help predict the location of the mutation. To some extent this is true, because significantly over modified type I collagen is predictive of a mutation in either the COL1A1 or COL1A2 gene. However, abnormal biochemistry cannot simply predict the location or type of mutation, and there are patients with normal biochemistry who nevertheless have OI, especially if they have one of the rare forms of autosomal recessive OI.
Linkage analysis can be performed in families (multiple affected members) with OI and can establish segregation to either COL1A1 or COL1A2. This technique is rarely used anymore owing to advances in molecular analyses. In the past, mutation detection in the type I collagen genes was an arduous and time-consuming task, but now has become much more standard of care, especially if identification of the mutation helps dictate clinical care or is used for reproductive choices (see the algorithm in Fig. 9.9). As previously noted, type I collagen biochemistry can be normal in some cases of OI, thus molecular analysis of type I collagen is the appropriate confirmative test if clinicians feel that they cannot completely rely on the well-established clinical and radiographic features of OI.
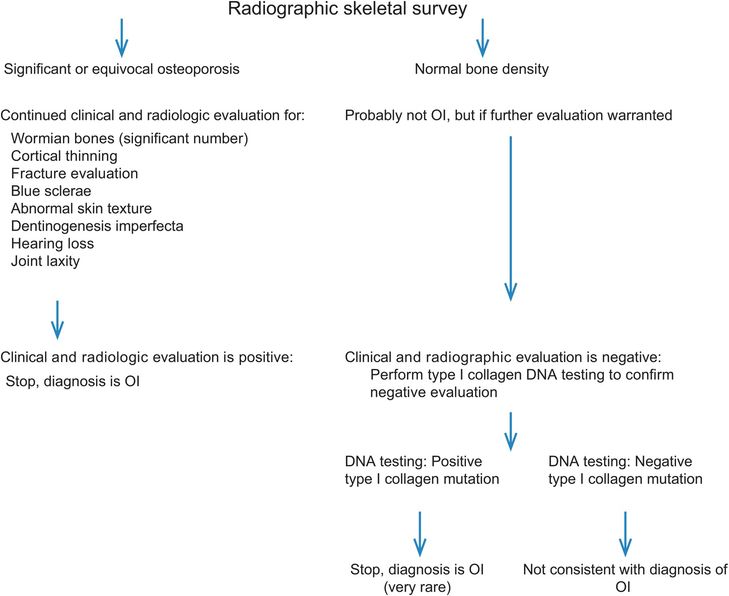
Figure 9.9 Algorithm to evaluate for OI in cases of suspected abuse.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


