Diffuse Lung Disease
Jeffrey S. Klein
Curtis E. Green
Diffuse lung disease represents a broad spectrum of disorders that primarily affect the pulmonary interstitium (Table 17.1). These diseases present in a variety of manners, most typically with symptoms of progressive dyspnea. Some patients, however, present with minimal or no symptoms and interstitial lung disease is discovered either incidentally or during radiologic screening for interstitial disease associated with collagen vascular disease. Restrictive lung disease and hypoxemia on pulmonary function tests are characteristically present. The radiographic findings produced by interstitial disease are reviewed in Chapter 12. Thin-section CT has revolutionized the diagnosis of interstitial lung disease, and its role in the evaluation of interstitial disease is detailed in this chapter.
Thin-Section Ct of the Pulmonary Interstitium
Normal Anatomy. Thin-section CT provides the most direct radiographic method for assessment of the pulmonary interstitium. The general utility of thin-section CT in the evaluation of chronic interstitial lung disease is outlined in Table 17.2 (1). The pulmonary interstitium is the scaffolding of the lung, providing support for the airways, gas-exchanging units, and vascular structures. It is a continuous network of connective tissue fibers that begins at the lung hilum and extends peripherally to the visceral pleura (see Fig. 12.10). The central interstitial compartment extending from the mediastinum peripherally and enveloping the bronchovascular bundles is termed the axial or bronchovascular interstitium. The axial interstitium is contiguous with the interstitium surrounding the small centrilobular arteriole and bronchiole within the secondary pulmonary lobule, where it is called the centrilobular interstitium. The most peripheral component of the interstitium is the subpleural or peripheral interstitium, which lies between the visceral pleura and the lung surface. Invaginations of the subpleural interstitium into the lung parenchyma form the borders of the secondary pulmonary lobules and represent the interlobular septa. Extending between the centrilobular interstitium within the lobular core and the interlobular septal/subpleural interstitium in the lobular periphery is a fine network of connective tissue fibers that support the alveolar spaces called the intralobular, parenchymal, or alveolar interstitium.
The secondary pulmonary lobule is defined as that subsegment of lung supplied by three to five terminal bronchioles and separated from adjacent secondary lobules by intervening connective tissue (interlobular septa) (Fig. 17.1). Each terminal bronchiole further subdivides into respiratory bronchioles, alveolar ducts, alveolar sacs, and alveoli. The unit of lung subtended from a single terminal bronchiole is called a pulmonary acinus. The centrilobular artery and preterminal bronchiole are located in the center of the secondary lobule. Pulmonary veins and lymphatics run at the margins of lobules within the interlobular septa, with lymphatics and connective tissue found within the contiguous subpleural interstitium. The secondary pulmonary lobule is typically polyhedral in shape, with each side ranging from 1 to 2.5 cm in length. The interlobular septa are most prominent over the periphery of the lung, where they are readily seen on CT. At the surface of the lung, these septa are short structures that course perpendicular to the pleural surface and completely separate adjacent lobules. Within the parahilar portions of the lung, the interlobular septa are longer and more obliquely oriented and incompletely marginate the secondary lobules.
Normal Thin-Section CT Findings. Thin-section CT can demonstrate much of the normal anatomy of the secondary pulmonary lobule. Interlobular septa are normally 0.1 mm thick and can be seen in the lung periphery, particularly along
the superior and inferior pleural surfaces (Fig. 17.2). Centrilobular arteries (1 mm in diameter) are V- or Y-shaped structures on thin-section CT seen within 5 to 10 mm of the pleural surface. Normal intralobular (0.7 mm) and acinar (0.3 to 0.5 mm) arteries are commonly seen. Normal airways are visible only to within 3 cm of the pleura. The centrilobular bronchiole, with a diameter of 1 mm and a wall thickness of 0.15 mm, is not normally visible on thin-section CT. Pulmonary veins (0.5 cm) are occasionally seen as linear or dot-like structures within 1 to 2 cm of the pleura and, when visible, indicate the locations of interlobular septa. The peribronchovascular, centrilobular, and intralobular interstitial compartments are not normally visible on thin-section CT.
the superior and inferior pleural surfaces (Fig. 17.2). Centrilobular arteries (1 mm in diameter) are V- or Y-shaped structures on thin-section CT seen within 5 to 10 mm of the pleural surface. Normal intralobular (0.7 mm) and acinar (0.3 to 0.5 mm) arteries are commonly seen. Normal airways are visible only to within 3 cm of the pleura. The centrilobular bronchiole, with a diameter of 1 mm and a wall thickness of 0.15 mm, is not normally visible on thin-section CT. Pulmonary veins (0.5 cm) are occasionally seen as linear or dot-like structures within 1 to 2 cm of the pleura and, when visible, indicate the locations of interlobular septa. The peribronchovascular, centrilobular, and intralobular interstitial compartments are not normally visible on thin-section CT.
Table 17.1 the Alphabet Soup of Interstitial Lung Disease | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
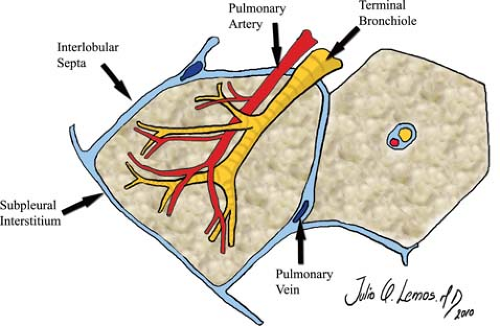 Figure 17.1. Diagram of the Normal Secondary Pulmonary Lobule. |
Table 17.2 Utility of Thin-Section Ct in the Evaluation of Chronic Interstitial Lung Disease | |
|---|---|
|
Thin-Section CT Signs of Disease
The signs of interstitial lung disease on thin-section CT are illustrated in Fig. 17.3 and their differential diagnosis is listed in Table 17.3 (1).
Interlobular (Septal) Lines. Septal thickening is most often seen as thin, short, 1- to 2-cm lines oriented perpendicular to and intersecting the costal pleura. These lines are best visualized in the subpleural and juxtadiaphragmatic regions of the lung, where they outline the anterior and posterior margins of secondary lobules. In the central regions of the lung, the thickened septa can completely envelope lobules to produce polygonal structures. Although septa can be seen in normal individuals, these lines are thicker (>1 mm) and more numerous in patients with diseases primarily affecting the interlobular interstitium, such as interstitial pulmonary edema, idiopathic pulmonary fibrosis (IPF), and lymphangitic carcinomatosis (Fig. 17.4). Interlobular lines on thin-section CT are the equivalent of Kerley B lines seen in the inferolateral portions of the lungs on frontal radiographs. Within the central regions of the lung, long (2 to 6 cm) linear opacities representing obliquely
oriented connective tissue septa are the equivalent of radiographic Kerley A lines.
oriented connective tissue septa are the equivalent of radiographic Kerley A lines.
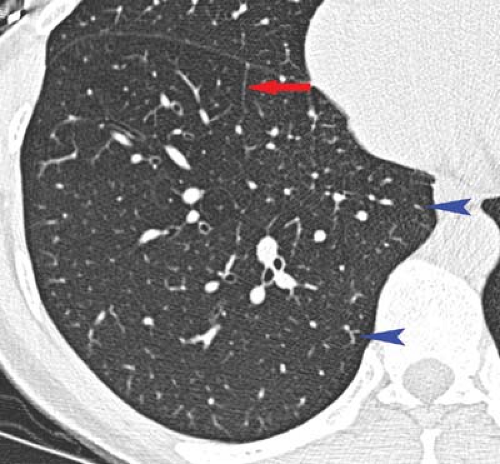 Figure 17.2. Thin-Section CT of Normal Lobular Anatomy. Normal interlobular septum (blue arrowheads) and centrilobular arteries (red arrow) are clearly visible. |
Table 17.3 Differential Diagnostic Thin-Section Ct Features in Interstitial Lung Disease | ||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||
Intralobular Lines. In some patients, a lattice of fine lines is seen within the central portion of the pulmonary lobule radiating out toward the thickened lobular borders to produce a “spoke-and-wheel” or “spider web” appearance. These lines are not normally visible on thin-section CT and represent thickening of the intralobular or parenchymal interstitium. Thickened intralobular lines usually result from fibrosis and are most commonly seen in idiopathic pulmonary fibrosis (IPF) and other causes of usual interstitial pneumonitis (UIP). Thickened intralobular lines can also be seen in other infiltrative diseases such as pulmonary alveolar proteinosis (PAP).
“Thickened” Fissures. The apparent thickening of interlobar fissures in patients with interstitial lung disease is usually a direct extension of the thickening of interlobular septa to involve the subpleural interstitium of the lung. While such a process normally involves all pleural surfaces equally, the “thickening” is usually best appreciated on the fissures, where two layers of visceral pleura—and therefore two layers of subpleural interstitium—are seen outlined on either side by aerated lung. The fissural thickening can be smooth or nodular. Smooth fissural thickening is virtually indistinguishable from a small amount of pleural fluid within the fissure and is most commonly seen with pulmonary edema. Nodular fissural thickening is commonly seen in sarcoidosis and lymphangitic carcinomatosis (Fig. 17.4), where the nodules lie within the subpleural lymphatics.
Thickened bronchovascular structures of the lung result from thickening of the peribronchovascular interstitium. This produces apparent enlargement of perihilar vascular structures and thickening of bronchial walls, which are the thin-section CT equivalent of peribronchial cuffing and tram tracking seen radiographically. Pulmonary edema causes smooth thickening of the peribronchovascular interstitium, whereas sarcoidosis causes nodular thickening. Lymphangitic carcinomatosis can
result in either smooth or irregular peribronchovascular thickening (Fig. 17.6).
result in either smooth or irregular peribronchovascular thickening (Fig. 17.6).
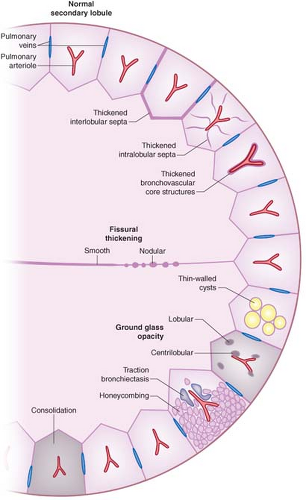 Figure 17.3. Thin-Section CT Findings in Interstitial Lung Disease (Adapted from The Radiologist, Baltimore: Williams & Wilkins, 1998, with permission). |
Centrilobular (Lobular Core) Abnormalities. Thickening of the axial interstitium within the lobular core produces an abnormal prominence of the “dot like” or branching centrilobular arteriole. Diseases that commonly produce this appearance include pulmonary edema, lymphangitic carcinomatosis, and UIP. The centrilobular bronchiole is not normally seen on thin-section CT but may be rendered visible as a result of luminal dilatation or thickening of the centrilobular interstitium. Small airways disease can produce centrilobular bronchiolar abnormalities, which are seen on thin-section CT as fluid-filled dilated branching Y-shaped structures that produce a “tree-in-bud” appearance. Ill-defined centrilobular nodules represent disease of the bronchiole and adjacent parenchyma and are commonly present in subacute hypersensitivity pneumonitis (Fig. 17.7), cryptogenic organizing pneumonia (COP), RB-ILD, as well as other disorders.
Subpleural Lines. These 5- to 10-cm-long curvilinear opacities are found within 1 cm of the pleura and parallel the chest wall. They are most frequent in the posterior portions of the lower lobes and remain unchanged on prone scans. They probably represent an early phase of lung fibrosis and should be distinguished from a similar line that is seen as a result
of atelectasis in the dependent portion of the lungs in normal individuals. Subpleural lines are most often seen in patients with asbestosis and, less commonly, IPF.
of atelectasis in the dependent portion of the lungs in normal individuals. Subpleural lines are most often seen in patients with asbestosis and, less commonly, IPF.
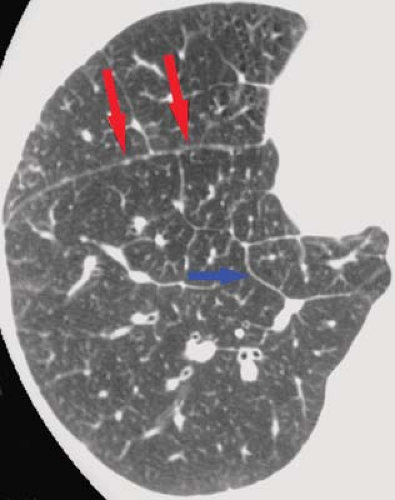 Figure 17.4. Interlobular (Septal) Lines in Lymphangitic Car cino ma tosis. A thin-section CT scan through the upper lobes in a patient with lymphangitic carcinomatosis shows thickened interlobular septa (blue arrow). Note the presence of nodular fissural thickening (red arrows), another common finding in this entity. |
Parenchymal bands are nontapering linear opacities, 2 to 5 cm in length, that extend from the lung to contact the pleural surface. These fibrotic bands can be distinguished from vessels and thickened septa by their length, thickness, course, absence of branching, and their association with regional parenchymal distortion. Parenchymal bands are frequently seen in asbestosis, IPF, and sarcoidosis.
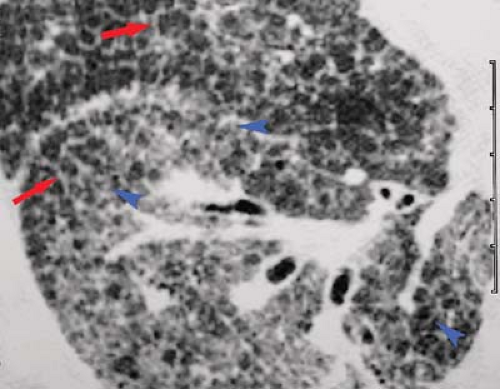 Figure 17.5. Intralobular Lines in Idiopathic Pulmonary Fibrosis (IPF). A targeted thin-section CT through the right lower lobe in a patient with IPF shows thickening of intralobular (red arrows) and interlobular (blue arrowheads) lines associated with ground glass opacity. |
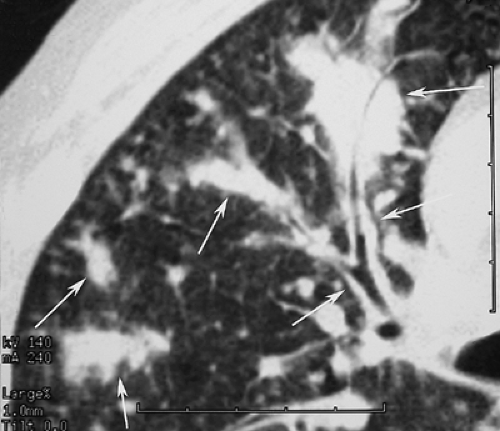 Figure 17.6. Thickened Bronchovascular Structures in Lymphangitic Carcinomatosis. In a patient with lymphangitic carcinomatosis, a thin-section CT shows both smooth and nodular thickening of the bronchovascular structures (arrows) that represents lymphatic tumor surrounding the axial interstitium. |
Honeycomb cysts are small (6 to 10 mm) cystic spaces with thick (1 to 3 mm) walls, most often in the posterior subpleural regions of the lower lobes and result from end-stage pulmonary fibrosis from a variety of etiologies. Pathologically, the cysts are lined by bronchiolar epithelium and are the result of bronchiolectasis. Most patients show additional signs of interstitial disease, including thickened interlobular and intralobular lines, parenchymal bands, irregularity of lung interfaces, and areas of ground glass opacity. Honeycombing is frequently seen in
UIP (Fig. 17.8) and chronic hypersensitivity pneumonitis and occasionally in sarcoidosis.
UIP (Fig. 17.8) and chronic hypersensitivity pneumonitis and occasionally in sarcoidosis.
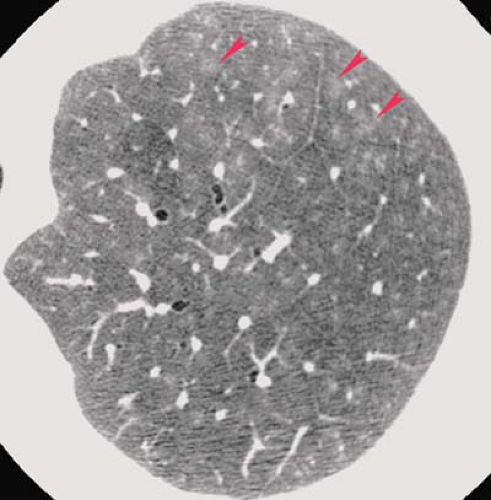 Figure 17.7. Centrilobular Ground Glass Nodules in Subacute Hypersensitivity Pneumonitis. Thin-section CT shows the typical poorly defined centrilobular nodules (arrowheads) of subacute hypersensitivity pneumonitis. |
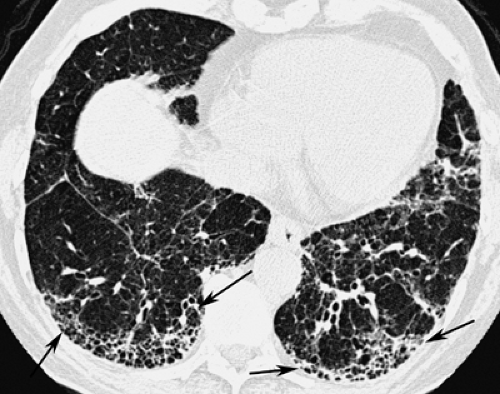 Figure 17.8. Honeycomb Lung in Idiopathic Pulmonary Fibrosis (IPF). Thin-section CT in a patient with IPF shows peripheral honeycombing (arrows) resulting from end-stage pulmonary fibrosis. |
Thin-walled cysts are a common manifestation of late stages of Langerhans’ cell histiocytosis of lung (LCH), also referred to as eosinophilic granulomatosis, and lymphangioleiomyomatosis (LAM). These cysts are slightly larger in diameter (10 mm) and have thinner walls than honeycomb cysts. Honeycomb cysts usually have shared walls, whereas the cysts of LCH and LAM do not. The cysts of LCH and LAM are usually evenly distributed from central to peripheral portions of the upper lobes (Fig. 17.9), with or without lower lobe involvement, whereas honeycombing tends to occur in the subpleural regions of the lower lobes. Normal lung may be found in the intervening spaces between the cysts of LCH and LAM. Honeycombing uniformly destroys lung and produces distortion of lung interfaces and traction bronchiectasis, features not typically found in either LCH or LAM.
Irregularity of Lung Interfaces. A common thin-section CT sign of interstitial disease, irregularity of the normally smooth interface between the bronchovascular bundles and the surrounding lung, reflects edema or fibrosis or infiltration of the axial interstitium by granulomas (Fig. 17.5) or tumor. Similarly, irregularity of the interface between fissures or pleural surfaces and adjacent lung indicates peripheral interstitial disease. UIP and sarcoidosis are the most common causes of irregular lung interfaces.
Micronodules. These 1- to 3-mm, sharply marginated, round opacities seen on thin-section CT represent conglomerates of granulomas or tumor cells within the interstitium. These are most often seen in sarcoidosis, LCH, silicosis (Fig. 17.10), miliary tuberculosis (TB) or histoplasmosis, metastatic adenocarcinoma, and lymphangitic carcinomatosis. They may be seen along the central bronchovascular structures (sarcoidosis, LCH), within interlobular septa or subpleural interstitium (sarcoidosis, lymphangitic carcinomatosis, silicosis), or within the substance of the pulmonary lobules (metastatic adenocarcinoma, miliary granulomatous infection). Nodules predominating in the peribronchovascular, interlobular, and subpleural regions—those portions of the interstitium where the lymphatics lie—are said to have a “perilymphatic” distribution.
Ground Glass Opacity. Ground glass opacity is defined as an area of increased attenuation (on thin-section CT) within which the normal parenchymal structures are visible. Multifocal areas of ground glass opacity can sometimes be identified in patients with diffuse interstitial lung disease. These regions often respect lobular borders and do not demonstrate air-bronchograms. They result from abnormalities below the resolution of the CT scan and are most often produced by thickening of the alveolar septa, with or without the lining of the alveolar spaces, by inflammatory exudate or fluid. Diseases commonly associated with this appearance include desquamative interstitial pneumonia (DIP), Pneumocystis jiroveci (formerly P carinii) pneumonia, acute hypersensitivity pneumonitis (Fig. 17.11), nonspecific interstitial pneumonia (NSIP), and interstitial pulmonary edema. The ground glass densities are occasionally confined to the immediate centrilobular regions of the pulmonary lobules, where they appear as fuzzy nodular densities that outline the normally invisible centrilobular bronchiole (Fig. 17.7). This reflects involvement of the peribronchovascular interstitium and surrounding
alveoli by an inflammatory process and is seen in hypersensitivity pneumonitis, COP, and panbronchiolitis. The presence of ground glass opacities is important because it often implies an active inflammatory process or edema that is reversible and warrants aggressive treatment. Ground glass abnormality associated with a predominant pattern of honeycombing indicates microscopic pulmonary fibrosis, however.
alveoli by an inflammatory process and is seen in hypersensitivity pneumonitis, COP, and panbronchiolitis. The presence of ground glass opacities is important because it often implies an active inflammatory process or edema that is reversible and warrants aggressive treatment. Ground glass abnormality associated with a predominant pattern of honeycombing indicates microscopic pulmonary fibrosis, however.
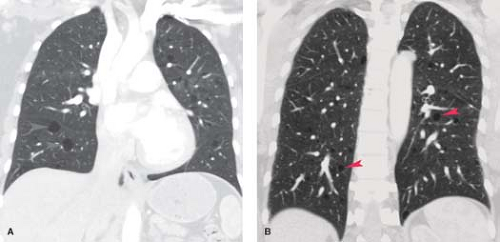 Figure 17.9. Thin-Walled Cysts in Lymphangioleiomyomatosis (LAM). A and B. Coronal reconstructions of a thin-section CT of a patient with LAM show multiple, thin-walled cysts (arrowheads). Although variably sized, the cysts are fairly uniform in shape. |
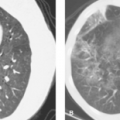
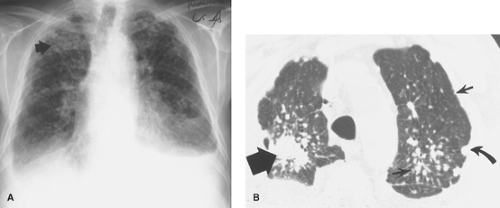 Figure 17.10. Nodules and a Conglomerate Mass in Silicosis. A. Posteroanterior radiograph of a 79-year-old patient with silicosis shows diffuse nodules as well as a conglomerate mass in the right upper lobe (arrow). B. Thin-section CT scan through the upper lobes shows peribronchovascular and subpleural micronodules (small arrows), larger nodules (curved arrow), and a conglomerate mass representing progressive massive fibrosis in the right upper lobe (large arrow). The pleural effusions are caused by concomitant congestive heart failure. |
Architectural Distortion and Traction Bronchiectasis. Processes that result in extensive parenchymal fibrosis can distort the normal architecture of the lung, creating irregularities of the lung–mediastinal, lung–pleural, and lung–vascular interfaces. Parenchymal distortion is often better appreciated on thin-section CT than on plain radiographs. Sarcoidosis and UIP (Fig. 17.12) are the diseases most commonly associated with architectural distortion.
A finding commonly associated with architectural distortion is traction bronchiectasis, in which fibrosis causes traction on the walls of bronchi, resulting in irregular dilatation. While this usually involves segmental and subsegmental bronchi, it also can be seen at the intralobular level, where traction bronchiolectasis contributes to honeycombing. Traction bronchiectasis is most commonly seen in UIP (Fig. 17.12), but is also common in fibrotic sarcoidosis and radiation fibrosis.
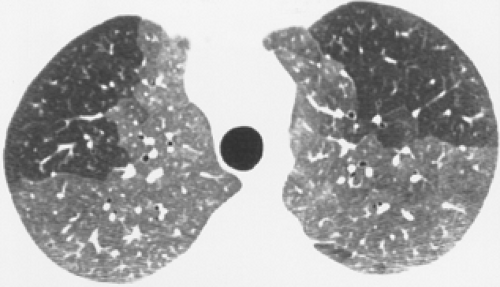 Figure 17.11. Ground Glass Opacity in Acute Hypersensitivity Pneumonitis. A thin-section CT through the upper lobes shows widespread ground glass opacity in a patient with hypersensitivity pneumonitis. Note that the pulmonary vessels are still visible within the areas of abnormality. |
Conglomerate Masses. In some patients with extensive pulmonary fibrosis, masses of fibrotic tissue develop in the parahilar regions of the upper lobes, often associated with peripheral bullae. On CT, these masses are seen to contain crowded vessels and dilated bronchi. They are most often seen in patients with end-stage sarcoidosis but can occur in complicated silicosis with progressive massive fibrosis (PMF) (Fig. 17.10) or radiation fibrosis following treatment of Hodgkin lymphoma or lung cancer. A similar finding is seen rarely in IV drug users when a granulomatous fibrosis results as a response to IV talc or starch mixed with narcotics.
Consolidation refers to increased lung density that obscures underlying blood vessels; air bronchograms are commonly present. This finding can be seen with any airspace-filling process (Fig. 17.13) but occasionally occurs in interstitial diseases such as UIP and sarcoidosis.
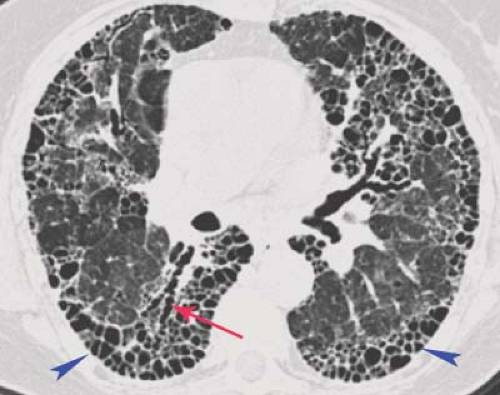 Figure 17.12. Architectural Distortion and Traction Bronchiectasis in Idiopathic Pulmonary Fibrosis. Thin-section CT through the lower lobes shows extensive peripheral honeycombing (blue arrowheads), traction bronchiectasis (red arrow), and architectural distortion. |
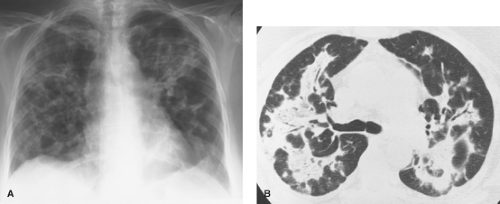 Figure 17.13. Consolidation in Cryptogenic Organizing Pneumonia (COP). A. Posteroanterior radiograph in a 53-year-old patient with fever, dyspnea, and a dry cough shows patchy consolidation and diminished lung volumes. B. Thin-section CT scan shows multifocal areas of consolidation in a peribronchial distribution. Note air bronchograms with mild bronchial dilatation (arrows) within the consolidated areas. An open lung biopsy showed COP. |
Chronic Interstitial Lung Disease
Chronic interstitial lung disease usually results from diffuse inflammatory processes that primarily affect the axial and parenchymal interstitium of the lung. A wide variety of disease processes can result in diffuse damage to the pulmonary interstitium (2). Careful evaluation of all available radiologic studies and correlation with clinical findings and laboratory data are essential to the accurate diagnosis of chronic interstitial lung disease (Table 17.4). However, the majority of patients with interstitial lung disease will require histologic examination of lung tissue for definitive diagnosis.
Chronic Interstitial Pulmonary Edema
Chronic elevation of pulmonary venous pressure may lead to increased interstitial markings on plain radiographs. The interstitial thickening is caused by distention of pulmonary lymphatics and chronic interstitial edema and is seen most commonly in patients with long-standing mitral stenosis or LV failure. Radiographically, peribronchial cuffing, tram tracking, poor definition of vascular markings, and linear or reticular opacities may be seen. Redistribution of blood flow to the upper lobes, a manifestation of pulmonary venous hypertension, and prominence of the fissures caused by subpleural edema and fibrosis are concomitant findings. Honeycombing is not a feature of chronic pulmonary venous hypertension; its presence in a patient with cardiac disease should suggest another cause of pulmonary fibrosis (e.g., amiodarone lung toxicity).
Connective Tissue Disease
These disorders are associated with immunologically mediated inflammation and damage to connective tissues throughout the body. The most common thoracic manifestations of this group of heterogeneous disorders are vasculitis and interstitial fibrosis, although the pleura, chest wall, diaphragm, and heart may also be affected (3).
Rheumatoid Lung Disease (Table 17.5). Rheumatoid arthritis produces a chronic arthritis of peripheral joints. Extra-articular manifestations are seen in up to 75% of patients. In contrast to the disease as a whole, which is more common in women, pulmonary involvement is more common in men. The pleuropulmonary manifestations of rheumatoid disease typically follow the onset of joint disease and tend to be seen in patients with high serum rheumatoid factor titers and eosinophilia, but in up to 15% of patients, pleuropulmonary involvement precedes the joint disease.
The most common radiographic manifestation of parenchymal lung involvement is an interstitial pneumonitis and fibrosis, which histologically is a form of UIP. This begins as an alveolitis (inflammation of the alveolar interstitium) that is seen radiographically as fine reticular or ground glass opacities with a lower zone predominance. There is gradual progression to end-stage pulmonary fibrosis with the development of a bibasilar medium or coarse reticular or reticulonodular pattern (honeycombing) (Fig. 17.14). Thin-section CT is more sensitive in detecting the earliest parenchymal changes than conventional radiographs and is also more sensitive in depicting the development of interstitial fibrosis (Fig. 17.15). Predominant upper lobe fibrosis, cavitation, and bulla formation are rare. This less common pattern of lung involvement is indistinguishable from that seen with ankylosing spondylitis and must be distinguished from postprimary fibrocavitary TB by acid-fast staining of sputum.
Less common parenchymal manifestations of rheumatoid disease are lung nodules (Fig. 17.15) and changes attributable to COP. Necrobiotic (rheumatoid) nodules in the lung can produce peripheral well-defined nodular opacities on chest radiographs that are histologically indistinguishable from the subcutaneous rheumatoid nodules seen on the extensor surfaces of the elbows and knees in these patients. The lung nodules commonly evolve into thick-walled cavities, which tend to wax and wane in parallel with the flares of arthritis. Similar nodules may develop in the lungs of coal miners and silica or asbestos workers with rheumatoid arthritis as a hypersensitivity response to inhaled dust particles (Caplan syndrome). Caplan syndrome is usually indistinguishable radiographically from the necrobiotic nodules of simple rheumatoid disease, although the presence of the associated characteristic small nodular or irregular parenchymal opacities of simple pneumoconiosis helps make this distinction. COP and bronchiolitis obliterans (constrictive bronchiolitis) are also associated with rheumatoid disease. The clinical, functional, and radiographic findings are similar to those of COP or bronchiolitis obliterans associated with systemic lupus erythematosus (SLE), drugs, or viral infection.
Pleuritis is the most common thoracic manifestation of rheumatoid disease and is found in 20% of patients. As with
pulmonary involvement, there is a male predilection for pleural disease. Pleural effusions are exudative and have a characteristically low glucose concentration.
pulmonary involvement, there is a male predilection for pleural disease. Pleural effusions are exudative and have a characteristically low glucose concentration.
Table 17.4 Differential Diagnostic Features in Chronic Interstitial Lung Disease | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Enlargement of the central pulmonary arteries and right heart dilatation may be seen on chest radiographs in patients with pulmonary arterial hypertension. This is an uncommon manifestation of rheumatoid disease that usually develops secondary to diffuse interstitial fibrosis. Rarely, the pulmonary arteries are involved as a part of the systemic vasculitis seen in extra-articular rheumatoid disease. There are no parenchymal abnormalities associated with rheumatoid pulmonary arteritis.
Abnormalities that may be seen in the chest wall of individuals with rheumatoid arthritis include tapered erosion of the distal clavicles, rotator cuff atrophy with a high-riding humeral head, bilateral symmetric glenohumeral joint space narrowing with or without superimposed degenerative joint disease, and superior rib notching or erosion.
Systemic Lupus Erythematosus (SLE). This disease of young and middle-aged women typically involves inflammation of multiple organs mediated by auto-antibodies and circulating immune complexes. The thorax is commonly affected and may be the initial site of involvement. The thoracic disease is often limited to the pleura and pericardium, although the lung, heart,
diaphragm, and intercostal muscles are involved in as many as one-third of patients. In the pleura and pericardium, a fibrinous serositis produces painful exudative pleural and pericardial effusions (see Fig. 19.5). Radiographically, the pleural effusions are small or moderate in size and can be unilateral or bilateral. The effusions usually resolve with corticosteroid therapy. Pleural fibrosis results in diffuse pleural thickening and is present in the majority of patients with long-standing disease.
diaphragm, and intercostal muscles are involved in as many as one-third of patients. In the pleura and pericardium, a fibrinous serositis produces painful exudative pleural and pericardial effusions (see Fig. 19.5). Radiographically, the pleural effusions are small or moderate in size and can be unilateral or bilateral. The effusions usually resolve with corticosteroid therapy. Pleural fibrosis results in diffuse pleural thickening and is present in the majority of patients with long-standing disease.
Table 17.5 Manifestations of Rheumatoid Lung Disease | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||
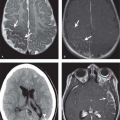
Pulmonary involvement may take the form of acute lupus pneumonitis or chronic interstitial disease. Acute lupus pneumonitis is characterized by rapid onset of fever, dyspnea, and hypoxemia and may require mechanical ventilation. These patients have pathologic changes that are indistinguishable from those seen in ARDS, with diffuse alveolar damage producing an exudative intra-alveolar edema with hyaline membrane formation. Radiographically, rapidly coalescent bilateral airspace opacities are seen, whereas the typical thin-section CT finding is one of ground glass opacity (Fig. 17.16). These findings are difficult to distinguish from those seen in diffuse alveolar hemorrhage associated with pulmonary vasculitis, severe pneumonia related to immunosuppressive therapy, or pulmonary edema secondary to renal failure. The diagnosis of acute lupus pneumonitis is made by excluding pneumonia and pulmonary edema and by noting an improvement following the initiation of immunosuppressive therapy.
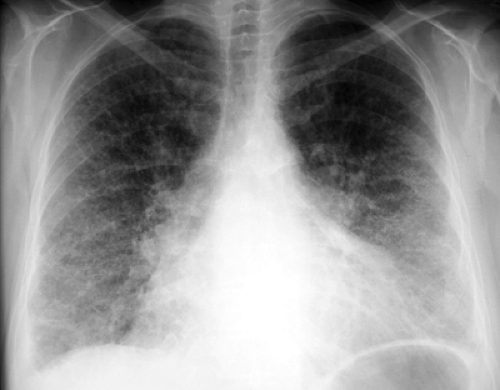 Figure 17.14. Honeycombing in Rheumatoid Lung. Posteroanterior radiograph in a patient with end-stage rheumatoid lung disease demonstrates medium reticular opacities caused by honeycomb cysts. Note the predominant peripheral distribution of disease. Bilateral pleural effusions and cardiac enlargement caused by pericardial effusion are also evident. |
Radiographic evidence of UIP is distinctly uncommon in SLE, but fibrosis is said to be present pathologically in one-third of patients. When seen radiographically, the pattern is one of bibasilar reticular opacities that are indistinguishable from those seen in rheumatoid lung disease or scleroderma. Therefore, the presence of severe interstitial fibrosis in a patient with clinical features of SLE should prompt consideration of the diagnosis of an overlap syndrome (mixed connective tissue disease). As with rheumatoid lung disease and scleroderma, thin-section CT is the most sensitive technique for demonstrating early interstitial disease.
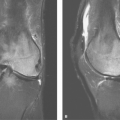
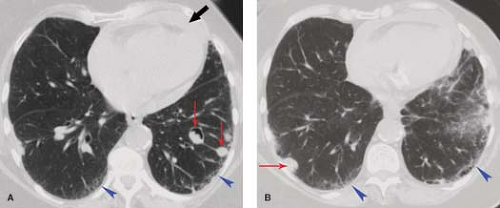 Figure 17.15. Rheumatoid Lung Disease and Rheumatoid Nodules. A and B. Thin-section axial CT scans through the lung bases in a patient with rheumatoid arthritis show asymmetric subpleural reticulation (blue arrowheads) reflecting interstitial pneumonitis. Note cavitating and solid bilateral rheumatoid nodules (red arrows) and pericardial effusion (black arrow), additional findings seen in patients with rheumatoid chest disease. |
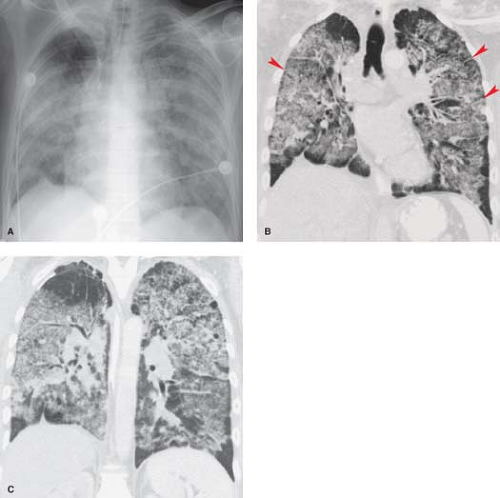 Figure 17.16. Acute Lupus Pneumonitis. A. Frontal chest radiograph in a 45-year-old woman with lupus and acute respiratory failure shows diffuse bilateral airspace and ground glass opacities. CT with coronal reformations through the mid- (B) and posterior (C) thorax shows bilateral ground glass opacities with foci of consolidation and associated interlobular septal thickening (arrowheads), findings seen in diffuse alveolar damage. |
Additional chest radiographic findings in SLE include elevation of the hemidiaphragms with decreased lung volumes and resultant bibasilar areas of linear atelectasis. Diaphragmatic elevation is present in as many as 20% of patients and is the result of diaphragmatic weakness from a primary myopathy unrelated to corticosteroid therapy. Rarely, the central pulmonary arteries are enlarged from pulmonary arterial hypertension secondary to pulmonary vasculitis. Pulmonary embolism with or without infarction may produce peripheral parenchymal opacities and results from deep venous thrombosis that develops in the presence of a circulating lupus anticoagulant. COP has been described in patients with SLE but is indistinguishable clinically and radiographically from lupus pneumonitis, because both conditions produce parenchymal opacities that are responsive to steroids. Superior rib erosions may be present and are indistinguishable from similar findings in rheumatoid arthritis or scleroderma.
Scleroderma (progressive systemic sclerosis) produces inflammation and fibrosis of the skin, esophagus, musculoskeletal system, heart, lungs, and kidneys in young and middle-aged women. The etiology and pathogenesis are unknown. The lungs are involved pathologically in nearly 90% of patients, although only 25% of patients have respiratory symptoms or radiographic evidence of pulmonary involvement. Pulmonary function testing is more sensitive than conventional radiography in the diagnosis of lung disease and shows the typical diminished lung volumes, preserved flow rates, and low diffusing capacity of interstitial pulmonary fibrosis. Pathologically, the parenchymal changes are those of interstitial fibrosis with a pattern of nonspecific interstitial pneumonia (NSIP). Severe pulmonary involvement is reflected radiographically as a coarse reticular or reticulonodular pattern involving the subpleural regions of the lower lobes. The most common thin-section CT findings are ground glass opacities, reticulation and eventually traction bronchiectasis in a lower zone, subpleural distribution (Fig. 17.17). Thin-section CT is more sensitive than the chest radiograph in detecting and evaluating interstitial disease. Progressive loss of lung volume is seen with advancing pulmonary fibrosis. The development of large (1 to 5 cm) subpleural lower lobe cysts may lead to spontaneous pneumothorax.
Pulmonary arterial hypertension with enlarged central pulmonary arteries and RV dilatation is seen in up to 50% of patients with scleroderma and may be seen in the absence of interstitial fibrosis. In these patients, thickening and obliteration of small muscular pulmonary arteries and arterioles are responsible for the development of pulmonary arterial hypertension. Pleural effusions are significantly less common in scleroderma than in rheumatoid disease or SLE and may be a
helpful distinguishing feature. Pleural thickening is more often attributable to extension of pulmonary interstitial fibrosis into the interstitial layer of the pleura than to pleuritis.
helpful distinguishing feature. Pleural thickening is more often attributable to extension of pulmonary interstitial fibrosis into the interstitial layer of the pleura than to pleuritis.
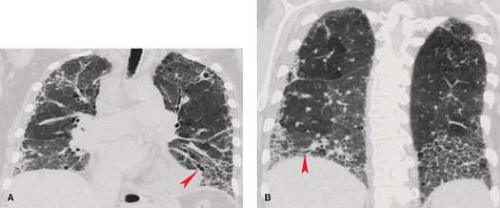 Figure 17.17. Scleroderma With Fibrotic Nonspecific Interstitial Pneumonitis (NSIP). CT with coronal reformations through the mid- (A) and posterior (B) thorax in a patient with biopsy-proven NSIP complicating scleroderma shows bilateral lower lobe predominant peripheral reticulation and ground glass opacities, with traction bronchiectasis (arrowheads) and minimal honeycomb cyst formation. |
Several additional chest radiographic findings may be seen in patients with scleroderma. Eggshell calcification of mediastinal lymph nodes has been reported, although it is more common in silicosis and sarcoidosis. A dilated air-filled esophagus may be identified on the upright chest radiograph and is a manifestation of esophageal dysmotility from smooth muscle atrophy and fibrosis. An air–fluid level within a dilated esophagus suggests secondary distal esophageal stricture formation from chronic reflux esophagitis. Functional or anatomic esophageal obstruction may result in aspiration with the development of lower lobe pneumonia. Because patients with scleroderma are at a greater risk for developing lung cancer, particularly bronchioloalveolar cell carcinoma, the appearance of a mass or persistent airspace opacity should raise this possibility. Patients with the CREST syndrome (subcutaneous calcification, Raynaud phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasia), a variant of scleroderma, may have radiographically visible calcifications within the subcutaneous tissues of the chest wall. Superior rib notching or erosion may also be seen.
Dermatomyositis and polymyositis involve autoimmune inflammation and destruction of skeletal muscle, producing proximal muscle pain and weakness (polymyositis) and occasionally associated with a skin rash (dermatomyositis). The thoracic manifestations of these diseases include respiratory and pharyngeal muscle weakness. Interstitial pneumonitis is seen in 5% to 10% of patients and is indistinguishable from that associated with rheumatoid lung disease, SLE, scleroderma, or IPF. A fine reticular interstitial pattern in acute disease leads to a chronic, coarse reticular or reticulonodular process that is predominantly basilar in distribution. Most patients with polymyositis and interstitial lung disease have clinical manifestations of rheumatoid arthritis or scleroderma, and these patients tend to respond favorably to corticosteroids. As with scleroderma, the early parenchymal changes may be subradiographic but can be demonstrated on thin-section CT studies through the lower lobes. Airspace consolidation and ground glass opacity representing organizing pneumonia and diffuse alveolar damage, respectively (Fig. 17.18). Additional chest radiographic findings in polymyositis reflect the involvement of skeletal muscle. Small lung volumes with diaphragmatic elevation and basilar linear atelectasis are secondary to diaphragmatic and intercostal muscle involvement. Pharyngeal and upper esophageal muscle weakness predispose to aspiration pneumonia. The chest radiograph should be examined carefully for lung masses because bronchogenic carcinoma accounts for a significant percentage of the malignancies seen with a higher-than-normal frequency in patients with dermatomyositis or polymyositis.
Sjögren Syndrome. This autoimmune disorder of middle-aged women is characterized by the sicca syndrome (dry eyes [keratoconjunctivitis sicca], dry mouth [xerostomia], and dry nose [xerorhinia]), which results from lymphocytic infiltration of the lacrimal, salivary, and mucous glands, respectively. Most patients with the sicca syndrome have associated manifestations of other collagen vascular diseases, such as rheumatoid arthritis, scleroderma, or SLE.
The chest is involved in approximately one-third of patients with Sjögren syndrome with or without associated collagen vascular disease. The most common manifestation is interstitial fibrosis, which is indistinguishable from that seen with other collagen vascular disorders. Involvement of tracheobronchial mucous glands leads to thickened sputum with mucus plugging and recurrent bronchitis, bronchiectasis, atelectasis, and
pneumonia. Thin-section CT demonstrates both interstitial opacities and the presence of small airways involvement with bronchiolectasis and a “tree-in-bud” appearance. Pleuritis and pleural effusion are less common.
pneumonia. Thin-section CT demonstrates both interstitial opacities and the presence of small airways involvement with bronchiolectasis and a “tree-in-bud” appearance. Pleuritis and pleural effusion are less common.
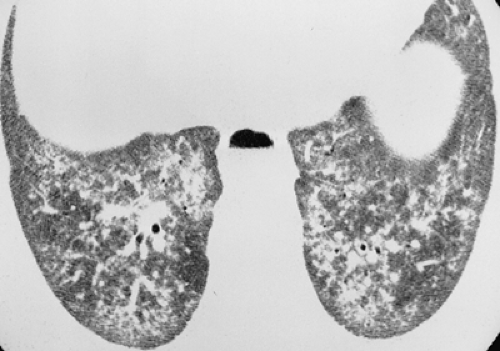 Figure 17.18. Polymyositis. Thin-section CT through the lung bases shows reticulation and ill-defined centrilobular nodules, likely reflecting interstitial pneumonitis and organizing pneumonia, respectively, in a patient with polymyositis.
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|