CHAPTER 62 Dilated Cardiomyopathy
Dilated cardiomyopathy (DCM) is the most common cardiomyopathy and is responsible for significant morbidity and mortality. The etiology of DCM is quite heterogeneous, and the DCM phenotype likely represents a common final outcome in response to many different myocardial insults; however, it has been recognized more recently that genetic factors probably account for 35% to 50% of all cases of DCM.1,2 Advances in imaging have provided insight into mechanisms of pathology in DCM, and have allowed more confident noninvasive separation of patients with ischemic DCM from patients with nonischemic DCM.
DEFINITION
DCM is a disease of the myocardium that is characterized by dilation and impaired systolic function of the left ventricle or both ventricles.3
PREVALENCE AND EPIDEMIOLOGY
DCM is the most common of the cardiomyopathies, accounting for approximately 55% of cases, and responsible for greater than 90% of cases referred to specialty centers.3 Idiopathic DCM is the most common cause of congestive heart failure in young patients with an estimated prevalence of 36.5 per 100,000 individuals in the United States, and is responsible for more than 10,000 deaths per year.4 Depending on the diagnostic criteria applied, the annual incidence in adults is 5 to 8 cases per 100,000 population.5 The true incidence is likely underestimated because many asymptomatic cases are unrecognized. DCM is responsible for a high proportion of cases of heart failure and sudden cardiac death, and is a leading cause of cardiac transplantation. The mortality rate in the United States owing to cardiomyopathy is greater than 10,000 deaths per year, with DCM the major contributor to this mortality.6
ETIOLOGY AND PATHOPHYSIOLOGY
DCM has been linked to many different etiologies, including infection, hypertension, pregnancy, alcohol, autoimmune disease, nutritional deficiency, cardiotoxins (e.g., anthracycline, heavy metals, cocaine, methamphetamines), genetic inheritance (e.g., mitochondrial disorders), or any cardiovascular disease in which the degree of myocardial dysfunction is not explained by the abnormal load conditions (e.g., valvular dysfunction) or the extent of ischemic damage.3 The World Health Organization definition of DCM excludes patients with enlarged and dysfunctional ventricles secondary to ischemic or valvular dysfunction, which are categorized as their own specific cardiomyopathy (i.e., ischemic cardiomyopathy and valvular cardiomyopathy).3 Viral myocarditis may be an important etiology in childhood, but in adults the relationship between myocarditis and DCM is less clear, and inconsistent results have been achieved in the attempt to isolate specific disease pathogens.
Family screening has emphasized the importance of inheritance in the etiology of DCM. Genetic transmission is most often autosomal dominant, with a lesser number of autosomal recessive, or X-linked, cases. Echocardiography family screening studies have shown abnormalities in approximately 25% of relatives of patients with DCM, including DCM and isolated left ventricular enlargement. Of individuals with left ventricular enlargement, 10% to 25% develop clinical DCM with symptomatic heart failure, arrhythmias, or thromboembolism within 5 years.7 These observations suggest that familial DCM is a slowly progressive disorder, and that screening of first-degree relatives of patients with DCM should assume a similar role to that of screening in hypertrophic cardiomyopathy patients, with more well-established genetic linkages.
The autosomal forms of familial DCM can be grouped into forms with a pure DCM phenotype or DCM with cardiac conduction system disease. Genetic heterogeneity is the hallmark of autosomal dominant DCM with 15 loci mapped for pure DCM and 5 for DCM with cardiac conduction system disease. These mutations include genes encoding cardiac actin, desmin, δ-sarcoglycan, β-sarcoglycan, cardiac troponin T, and α-tropomyosin. Most genes identified to date encode either cytoskeletal or sarcomeric proteins. These proteins are important for structural integrity and for force transmission.2
X-linked DCM occurs in adolescent boys and young men with rapid progression from congestive heart failure to death or transplantation, and is characterized by mutations in the gene for cardiac dystrophin, a cytoskeletal protein providing structural support to the myocyte and linking it to the sarcolemma. The dystrophin gene is also responsible for Duchenne and Becker muscular dystrophies, which also have DCM as a prominent feature.8
Histologic changes associated with DCM are frequently nonspecific, and not all features may be present. DCM is characterized by progressive interstitial fibrosis with a reduced number of functional myocytes and, in advanced stages, relative wall thinning. Although atrophic changes predominate histologically, there is also myocyte elongation with an addition of newly formed sarcomeres, which is the major factor responsible for increased chamber size. Myocyte diameter increases, but is inadequate to preserve a normal ratio of wall thickness to chamber diameter. Myocyte nuclear hypertrophy and pleomorphism may also be seen. There is often an increase in interstitial T lymphocytes and focal accumulation of macrophages associated with individual myocyte death.8
MANIFESTATIONS OF DISEASE
Imaging Technique and Findings
Ultrasonography
Echocardiography reveals a dilated left ventricular cavity with reduced global function. End-systolic and end-diastolic diameter are increased, and ejection fraction, fractional shortening, stroke volume, and cardiac output are decreased (Figs. 62-1 and 62-2).
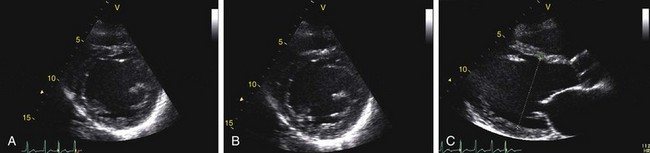
 FIGURE 62-1
FIGURE 62-1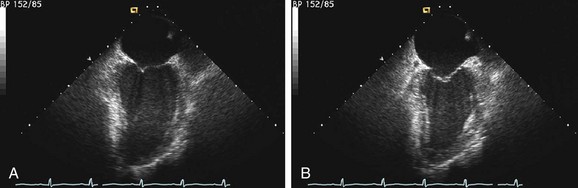
 FIGURE 62-2
FIGURE 62-2Evaluation of right ventricular function is also important. Patients with biventricular dysfunction have a lower New York Heart Association functional class, tend to have more severe left ventricular dysfunction, and have a worse long-term prognosis.9,10
Stress echocardiography may also play a role in assessment of patients with DCM. Patients with improvement in ejection fraction greater than 20% during stress echocardiography have a better prognosis. Drozd and colleagues11 showed that the incidence of cardiac death or transplantation is lower in patients with preserved contractile reserve. Another study assessed the prognostic significance of high-dose dobutamine stress echocardiography, and concluded that the change in wall motion score index is able to identify patients at greater risk for cardiac death during follow-up, and that change in wall motion score index had superior prognostic information to change in ejection fraction.12
Stress echocardiography with dipyridamole has also been used to assess coronary flow reserve in patients with DCM. Rigo and colleagues13 evaluated 129 patients with DCM and found that coronary flow reserve, assessed via Doppler velocity interrogation of the mid left anterior descending coronary artery, was often impaired, and that reduced coronary flow reserve was an independent marker of poor prognosis. Pratali and coworkers14 performed dobutamine and dipyridamole stress echocardiography in 87 patients with DCM and found that both tests have similar feasibility and prognostic accuracy.



