Embryology
At the end of the third week of gestation, the embryo, a relatively flat disk, begins to form a tube by means of four folds. These include one cephalic, one caudal, and two lateral folds, which combine to form the anterior abdominal wall at the region of the umbilicus. Failure of these folds to completely unite may result in defects of the anterior abdominal wall. The type of defect created depends on which of the folds has failed to fuse.
In the developing embryo, the midgut, the portion of bowel from the insertion of the bile duct to the splenic flexure of the colon, rapidly elongates at approximately the fifth to sixth week of gestation, and the abdominal cavity transiently becomes too small to accommodate this bowel. This results in physiologic herniation of bowel into the extraembryonic celom in the umbilical cord. These herniated loops of bowel return to the abdomen at approximately the 10th week of gestation. During this time, the bowel undergoes a 270-degree counterclockwise rotation. The failure of the bowel to return to the abdominal cavity results in an omphalocele. Gastroschisis occurs when herniation of abdominal viscera occurs through a defect in the anterior abdominal wall, usually to the right of the umbilicus.
The increasing use of prenatal ultrasound and routine screening with maternal serum α-fetoprotein has enabled earlier diagnosis of defects of the anterior abdominal wall, including pentalogy of Cantrell, omphalocele, gastroschisis, and bladder exstrophy. Although effort has been made to find a common cause for these abdominal wall defects, they seem to be disparate entities. Overall, the prognosis for these conditions tends to depend on associated congenital anomalies.
Defects of the Anterior Abdominal Wall
Pentalogy of Cantrell
Pentalogy of Cantrell is composed of two major abnormalities—ectopia cordis and a midline thoracoabdominal wall defect—associated with abnormalities of the tissues between these two areas and with defects of the lower sternum, diaphragmatic pericardium, and anterior diaphragm. Less severe cases have been described, which are considered to be incomplete forms. The cause is unclear, but there may be a genetic component. Numerous other abnormalities are associated with pentalogy of Cantrell, including cleft lip and palate and limb anomalies.
Clinically, patients generally present with dyspnea and cyanosis. These newborns have an anterior abdominal wall defect, usually an omphalocele, and a structural heart defect, such as atrial septal defect, ventricular septal defect, or tetralogy of Fallot. The anterior abdominal wall defect, paired with the short or cleft sternum, gives the appearance of a high epigastrium filled by the heart.
Diagnosis of pentalogy of Cantrell is usually made by prenatal ultrasound, when findings of omphalocele, ectopia cordis, and congenital heart disease are confirmed. Postnatally, chest radiographs demonstrate abnormal positioning of the heart, usually with dextrorotation. Thoracic abnormalities may also be evident, including pulmonary hypoplasia and rib anomalies. Computed tomography (CT) can be useful in preoperative evaluation of the thoracic cavity before repair of ectopia cordis. CT angiography and magnetic resonance imaging (MRI) can define cardiovascular anatomy, the degree of diaphragmatic deficiency, and the degree of transdiaphragmatic herniation of the bowel or liver.
Treatment of pentalogy of Cantrell involves closure of the thoracoabdominal wall defect, but success with corrective surgery has been limited. Postoperative complications include increased intrathoracic and intra-abdominal pressures, which can result in respiratory and cardiovascular compromise. The prognosis depends on the severity of the cardiac anomalies and on the severity of any other associated abnormalities.
Omphalocele
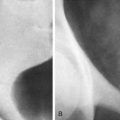
An omphalocele is herniation of abdominal viscera into the intact umbilical cord, with a membranous covering over the eviscerated organs unless there has been membrane rupture ( Fig. 123-1 ). The spectrum of severity ranges from a small umbilical hernia to a large defect, resulting in evisceration of all abdominal organs. The incidence of omphalocele is 1 case in 4000 to 7000 live births. Although an omphalocele can be an isolated abnormality, associated anomalies are more common with an omphalocele than with gastroschisis. Associated genetic abnormalities are seen in up to 54% of patients; trisomy 18 is most common, but trisomy 13, Beckwith-Wiedemann syndrome, and rarely uniparental disomy (i.e., inheritance of both copies of a chromosome pair from one parent and none from the other) also occur. Studies suggest that associated chromosomal abnormalities are more likely in patients with a small omphalocele that does not include herniated liver. Visceral abnormalities are seen in up to 70%, including neural tube defects and cardiac, renal, facial, and skeletal abnormalities and pentalogy of Cantrell. Gastrointestinal anomalies can include imperforate anus, colonic atresia, and Hirschsprung’s disease.

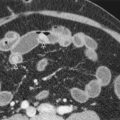
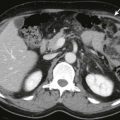
Physiologic herniation of bowel is normally seen between the 8th and 12th weeks of gestation; omphalocele cannot be reliably diagnosed until after this period. Omphalocele usually is diagnosed on prenatal ultrasound; patients with omphalocele demonstrate abdominal viscera within a mass at the umbilical cord insertion site with a covering membrane ( Fig. 123-2 ). The abdominal wall defect of an omphalocele tends to be large. The herniated viscera usually include liver with various amounts of bowel. When the omphalocele contains herniated liver and biliary structures, it is referred to as a giant omphalocele ( Fig. 123-3 ). Ascites can be seen within the abdomen or omphalocele, and polyhydramnios may be present. Prenatal diagnosis should also include a thorough search for associated anomalies. Fetal MRI is increasingly performed in patients with omphalocele to characterize the size and location of the anterior wall defect, to characterize the sac and its contents, and to identify associated anomalies (see Fig. 123-3 ). In addition, fetal MRI can be used to calculate lung volumes in patients with giant omphalocele to predict the degree of pulmonary hypoplasia, which is predictive of postnatal morbidity. Postnatal imaging of infants with omphalocele is generally reserved for identification of associated anomalies and should include echocardiography and renal ultrasound.


Studies of the route of delivery for infants with omphalocele have demonstrated no significant improvement in outcome with cesarean delivery compared with vaginal delivery. Cesarean delivery usually is reserved for patients with obstetric indications. Exceptions are patients who have a giant omphalocele, who are at increased risk of fatal hemorrhage from liver injury.
Unlike in gastroschisis, the herniated bowel and liver in an omphalocele are morphologically and functionally normal. The membrane that covers the herniated viscera in an omphalocele decreases fluid loss and protects against the metabolic abnormalities commonly seen in patients with gastroschisis.
The treatment of omphalocele is dictated by the presence of comorbidities (cardiovascular compromise, pulmonary insufficiency), the coexisting congenital syndromes or anomalies, and the size of the defect. Surgical options include primary closure for smaller defects and delayed closure, by the “paint and wait” technique, allowing time for the patient to grow enough to accommodate larger omphaloceles (see Fig. 123-1 ). Functionally, patients with omphalocele are typically spared the problems of bowel motility associated with gastroschisis. The protective covering membrane over the omphalocele affords a protective barrier to amniotic fluid exposure. The prognosis of patients with omphalocele predominantly depends on associated chromosomal or structural anomalies. Infants with an omphalocele and a cardiac anomaly have a mortality rate approaching 80%. Prognosis is poorer when the covering membrane has ruptured because these patients tend to have significantly smaller birth weight than those with an intact membrane, regardless of omphalocele size. Patients with giant omphaloceles also tend to have pulmonary hypoplasia, which can complicate primary repair.
Gastroschisis
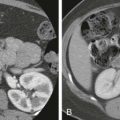
Gastroschisis refers to herniation of abdominal viscera, usually the small and large bowel, through a defect in the anterior abdominal wall, typically to the right of the umbilicus. The umbilical cord insertion is intact, and there is no membrane covering the eviscerated organs ( Fig. 123-4 ). These bowel loops are subsequently exposed to amniotic fluid in utero.

The incidence of gastroschisis is 1 case in 10,000 live births, but epidemiologic studies from the United States, Europe, and Japan have found increasing rates of gastroschisis, by as much as 10-fold, during the past decade. This is not thought to represent improved detection or reporting because the incidence of omphalocele has remained stable during the same period, and the rate at which the incidence has increased suggests environmental risk factors rather than genetic ones. Although reports of familial gastroschisis suggest an inherited propensity, numerous studies have suggested that teratogens play a role. Gastroschisis is more likely to occur in young mothers who are heavy smokers, use alcohol during pregnancy, have poor nutrition during pregnancy, or use over-the-counter medications with vasoactive properties (e.g., acetaminophen, aspirin, pseudoephedrine, phenylpropanolamine, ephedrine, methylenedioxymethamphetamine [ecstasy]) early in pregnancy. Clusters of infants with this defect have also been described near toxic waste sites.
Unlike omphalocele, gastroschisis tends not to be associated with chromosomal abnormalities. Up to 31% of male patients with gastroschisis have cryptorchidism, and the testicles may be part of the herniated viscera. Up to 25% of patients with gastroschisis have intestinal atresia or stenosis or other bowel complications. Other than bowel atresias and cryptorchidism, associated anomalies are rare.
The cause of intestinal atresia with gastroschisis is unclear, with theories suggesting a common vascular insult causing the gastroschisis and atresia rather than bowel ischemia from constriction by the defect in the anterior abdominal wall. In addition to atresia, the herniated bowel in patients with gastroschisis is usually damaged, with bowel shortening, thickening, and development of a fibrous covering. Most patients have problems with poor bowel motility and absorptive function long after surgical correction of the defect. Studies have suggested that the causes include exposure to amniotic fluid, constriction-induced injury at the defect, and altered gene expression within enterocytes. The bowel injury that occurs in utero seems to occur late in pregnancy, with findings of progressive bowel dilation and thickening. Asymmetric bowel wall dilation, with or without meconium peritonitis, suggests bowel atresia. Not all bowel injury occurs in utero; improved prognosis has been described for early repair or silo application, suggesting that some damage may occur postnatally.
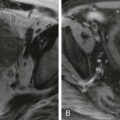
The diagnosis of gastroschisis is typically made on prenatal ultrasound, and maternal screening demonstrates elevated serum levels of α-fetoprotein. The diagnosis of gastroschisis can be made as early as 12 weeks of gestation by ultrasonographic identification of herniated viscera without a covering membrane in the presence of a normal umbilical cord insertion ( Fig. 123-5 ). The abdominal wall defect is usually small, most often less than 4 cm in diameter. In addition to bowel, organs that can herniate include liver, stomach, testes, and fallopian tubes.

Prenatal diagnosis enables early diagnosis of other abnormalities and allows decisions to be made about the location and timing of delivery, optimizing postnatal care. As with omphalocele, studies have failed to show improved survival or better outcomes for infants with gastroschisis delivered by cesarean section compared with vaginal delivery, with the exception of cases with eviscerated liver. Infants delivered at medical centers with postnatal and surgical experience in the care of gastroschisis have improved outcome compared with patients delivered elsewhere.
Treatment of gastroschisis involves the return of the herniated organs to the abdominal cavity with primary closure, if possible. This tends to be more urgent with gastroschisis than with omphalocele because the lack of a covering membrane results in increased fluid loss. If the abdominal cavity is too small to tolerate a primary repair because of size, a silo repair may be necessary, with the gradual return of the eviscerated organs to the abdominal cavity during several days (see Fig. 123-4 ). One risk of closure includes increased intra-abdominal pressures that can cause vascular and respiratory compromise, urethral obstruction, and bowel ischemia. In cases of cryptorchidism, the testicles are usually returned to the abdominal cavity at the time of repair of the abdominal wall defect, and most eventually descend to the normal scrotal position.
The prognosis for patients with gastroschisis depends on the presence of small bowel atresia or stenosis and on the condition of the herniated bowel. Postnatal imaging may be requested to detect small bowel strictures or areas of perforation, both of which are seen in complex gastroschisis and may not be predicted by prenatal imaging ( Fig. 123-6 ). Although the cause of bowel injury is unclear, it affects outcome by leading to intrauterine growth retardation, amniotic fluid abnormalities, and preterm labor. Postnatally, the bowel injury results in poor weight gain, decreased bowel motility, and feeding intolerance. Return of bowel motility is slow, regardless of the presence or absence of bowel atresia or stenosis.

Mortality of patients with gastroschisis is commonly related to short gut, which can be congenital or can result from volvulus or necrotizing enterocolitis. Mortality can also be tied to liver disease from prolonged parenteral nutrition, particularly in patients with bowel atresia.
Exstrophy of the Bladder
With an incidence of approximately 2 cases per 100,000 live births, bladder exstrophy is a rare congenital anomaly. Epidemiologic studies suggest an equal male-to-female ratio and a higher incidence among white patients compared with nonwhite patients (e.g., blacks, Hispanics, Asians).
Bladder exstrophy results from a failure of closure of the anterior abdominal wall at the ventral end of the cloacal membrane. This defect produces anterior herniation of the bladder, bladder neck, and urethra. There is also malformation of the external genitalia, including epispadias and a small phallus in male patients and an open urethral plate and labial separation in female patients. Other associated anomalies can include cleft palate, neural tube defects, cardiovascular and musculoskeletal abnormalities, and preterm birth.
Because the findings of bladder exstrophy can be subtle, the diagnosis is often not made on the basis of routine screening prenatal ultrasound. Persistent nonvisualization of the bladder in combination with normal-appearing kidneys and a normal volume of amniotic fluid should raise the suspicion of bladder exstrophy. Care must be taken to avoid mistaking a completely empty bladder for an absent one because the fetal bladder normally fills and empties every 50 to 155 minutes. A soft tissue mass that can be seen in the lower abdominal wall represents the exstrophied bladder. Additional findings of bladder exstrophy include a low insertion of the umbilical cord, a small phallus and epispadias in male patients, and splaying of the iliac crests.
Bladder exstrophy repair usually is staged. The initial stage includes closure of the bladder, posterior urethra, and anterior abdominal wall, possibly with pelvic osteotomy, depending on the amount of pubic diastasis present at birth. Subsequent stages include epispadias repair and bladder neck reconstruction. Findings suggest greater success when bladder closure is performed early.
As with other defects of the anterior abdominal wall, there has been no evidence to suggest that cesarean section improves outcome, but delivery at a hospital with experience in surgical care of bladder exstrophy is recommended because of resultant improved outcomes. There is no significant perinatal mortality associated with bladder exstrophy; however, there can be significant long-term morbidity, particularly urinary, with approximately 70% of patients requiring major reconstruction to achieve continence. Male and female patients experience decreased fertility.
Defects of the Anterior Abdominal Wall
Pentalogy of Cantrell
Pentalogy of Cantrell is composed of two major abnormalities—ectopia cordis and a midline thoracoabdominal wall defect—associated with abnormalities of the tissues between these two areas and with defects of the lower sternum, diaphragmatic pericardium, and anterior diaphragm. Less severe cases have been described, which are considered to be incomplete forms. The cause is unclear, but there may be a genetic component. Numerous other abnormalities are associated with pentalogy of Cantrell, including cleft lip and palate and limb anomalies.
Clinically, patients generally present with dyspnea and cyanosis. These newborns have an anterior abdominal wall defect, usually an omphalocele, and a structural heart defect, such as atrial septal defect, ventricular septal defect, or tetralogy of Fallot. The anterior abdominal wall defect, paired with the short or cleft sternum, gives the appearance of a high epigastrium filled by the heart.
Diagnosis of pentalogy of Cantrell is usually made by prenatal ultrasound, when findings of omphalocele, ectopia cordis, and congenital heart disease are confirmed. Postnatally, chest radiographs demonstrate abnormal positioning of the heart, usually with dextrorotation. Thoracic abnormalities may also be evident, including pulmonary hypoplasia and rib anomalies. Computed tomography (CT) can be useful in preoperative evaluation of the thoracic cavity before repair of ectopia cordis. CT angiography and magnetic resonance imaging (MRI) can define cardiovascular anatomy, the degree of diaphragmatic deficiency, and the degree of transdiaphragmatic herniation of the bowel or liver.
Treatment of pentalogy of Cantrell involves closure of the thoracoabdominal wall defect, but success with corrective surgery has been limited. Postoperative complications include increased intrathoracic and intra-abdominal pressures, which can result in respiratory and cardiovascular compromise. The prognosis depends on the severity of the cardiac anomalies and on the severity of any other associated abnormalities.
Omphalocele
An omphalocele is herniation of abdominal viscera into the intact umbilical cord, with a membranous covering over the eviscerated organs unless there has been membrane rupture ( Fig. 123-1 ). The spectrum of severity ranges from a small umbilical hernia to a large defect, resulting in evisceration of all abdominal organs. The incidence of omphalocele is 1 case in 4000 to 7000 live births. Although an omphalocele can be an isolated abnormality, associated anomalies are more common with an omphalocele than with gastroschisis. Associated genetic abnormalities are seen in up to 54% of patients; trisomy 18 is most common, but trisomy 13, Beckwith-Wiedemann syndrome, and rarely uniparental disomy (i.e., inheritance of both copies of a chromosome pair from one parent and none from the other) also occur. Studies suggest that associated chromosomal abnormalities are more likely in patients with a small omphalocele that does not include herniated liver. Visceral abnormalities are seen in up to 70%, including neural tube defects and cardiac, renal, facial, and skeletal abnormalities and pentalogy of Cantrell. Gastrointestinal anomalies can include imperforate anus, colonic atresia, and Hirschsprung’s disease.

Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


