Imaging Modalities
Ultrasound is typically the first imaging modality used in the evaluation of children with suspected biliary disease because it is noninvasive and relatively inexpensive and does not involve contrast agents, sedation, or radiation exposure. Ultrasound is particularly useful in differentiating obstructive from nonobstructive causes of jaundice. In general, the only preparation needed for an ultrasound study in the work-up of suspected biliary disease is for the child to have nothing by mouth for as close to 4 to 6 hours as possible to ensure optimal distention of the biliary system.
Computed tomography (CT) and magnetic resonance imaging (MRI) can also be useful in the evaluation of the biliary system in children, but CT exposes the child to radiation and both imaging modalities may require sedation. Specific protocols for evaluating the biliary system have been developed for CT and MRI. CT cholangiography, performed after the intravenous administration of a contrast agent that is excreted by the liver into the biliary system (meglumine iodoxamic acid), is not typically used in the pediatric population. Magnetic resonance cholangiopancreatography (MRCP) is a noninvasive means of evaluating the biliary system, the surrounding liver parenchyma, and other abdominal viscera that can usually be performed without administration of a contrast agent and does not expose the child to radiation. MRCP is limited in neonates and infants, however, as biliary ducts less than 1 mm in diameter are difficult to visualize on MRCP. In preparation for MRCP, it is essential that the child be given nothing by mouth for as close to 4 to 6 hours as possible so that the biliary system is optimally distended and the stomach is empty. During acquisition of the images for MRCP, breath-hold techniques can be used in children who are able to cooperate, and non–breath-hold techniques with respiratory gating can be used in children who are not able to cooperate.
Percutaneous transhepatic cholangiography, percutaneous cholecystocholangiography, intraoperative cholangiography, and endoscopic retrograde cholangiopancreatography (ERCP) are also used in evaluation of the pediatric biliary system, but they are invasive examinations, expose the child to radiation and sedation, and can be technically challenging. ERCP, for example, has a complication rate in pediatric patients of one in three (33%), which is higher than the complication rate for adults.
Nuclear medicine hepatobiliary scintigraphy, which is performed with either 99m Tc-disofenin ([2,6-diisopropylacetanilido]-iminodiacetic acid) or 99m Tc-mebrofenin (bromo-2,4,6-trimethylacetanilidoiminodiacetic acid), provides more physiologic information than anatomic information, particularly regarding the patency of the biliary system.
Normal Gallbladder
Ultrasound is an ideal imaging modality for evaluating the gallbladder in children because it allows real-time evaluation of the gallbladder in multiple planes and with the patient in various positions. The normal pediatric gallbladder is approximately 1.3 cm in length in neonates and 3.4 cm in length in infants and less than 1 cm in width. The average length of the gallbladder in a 16-year-old is 8 cm, with an average width of less than 3.5 cm.
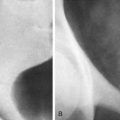
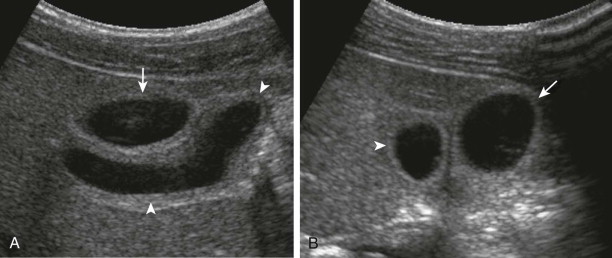
The normal sonographic appearance of the gallbladder wall is well defined, hyperechoic, and less than 3 mm in thickness when the child is fasting. Duplication of the gallbladder is rare and is often asymptomatic, but when it is encountered, it may be mistaken for a pathologic process such as choledochal malformation, dilated bile ducts, or gallbladder diverticulum ( Fig. 119-1 ). Other congenital anomalies of the gallbladder include ectopia (most commonly retrohepatic, intrahepatic, or suprahepatic), septations, and agenesis. The normal pediatric gallbladder may have folds or kinks, which can mimic pathologic processes such as stones and dilated bile ducts. Imaging in more than one plane or with the patient in different positions can help differentiate normal folds from abnormal pathologic processes ( Fig. 119-2 ).

Gallbladder wall thickening in children, which is defined as more than 3 mm in thickness, can be seen in the setting of acute or chronic cholecystitis, hypoalbuminemia, ascites, systemic venous hypertension, and acute hepatitis. Children with ascites and hypoalbuminemia can have normal gallbladder wall thickness, however. Cholecystitis can occur in children without gallbladder wall thickening as well. The gallbladder wall may appear falsely thickened if fluid is trapped in the mesentery between the gallbladder and liver, creating a halo effect, or if fluid is otherwise surrounding the intraperitoneal gallbladder. Technical factors, such as the angle of the transducer, can also affect the appearance of the gallbladder wall. If the gallbladder is not optimally distended because of a contraction from a recent meal, the gallbladder wall may appear falsely thickened, and repeated imaging after the patient has been fasting for 4 to 6 hours is recommended for more accurate evaluation.
Normal Bile Ducts
Bile excreted by hepatocytes of the liver is collected by bile canaliculi that merge into the canals of Hering. Bile then flows into interlobular bile ducts, more peripheral intrahepatic bile ducts, and then into the left and right hepatic ducts. The left and right hepatic ducts merge into the common hepatic duct, which exits the liver and joints the cystic duct (from the gallbladder), which then forms the common bile duct. The common bile duct then joins the pancreatic duct, forming the ampulla of Vater, which delivers the bile to the second portion of the duodenum.
Peripheral intrahepatic ducts are not typically visualized on ultrasound, CT, or MRI (even MRCP) unless they are dilated. The normal left and right hepatic ducts may be seen on ultrasound, MRI, and CT in older children. The common hepatic duct, cystic duct, and common bile duct may be visualized more readily, but their small size can make visualization challenging. As measured on ultrasound from inner wall to inner wall, the diameter of the common bile duct should be less than 1.6 mm in children younger than 1 year and less than 3.3 mm during childhood and early adolescence. The cystic duct is usually seen only if it is dilated, and even then, only the distal portion of the cystic duct is typically visualized.
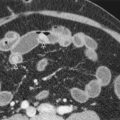
On MRCP, the pediatric biliary tree is considered normal if the right hepatic, left hepatic, common hepatic, and common bile ducts are visualized, are of normal caliber, and have confluent, uniform branching ( Fig. 119-3 ). In neonates, however, the extrahepatic biliary ducts may be the only ducts visible.

Evaluation of Infants and Children with Cholestasis
Neonatal Jaundice
Normal transient physiologic jaundice (unconjugated hyperbilirubinemia) can occur in term and preterm infants in the first 2 weeks of life. The Cholestasis Guideline Committee recommends evaluation for cholestasis (abnormal accumulation of conjugated bilirubin), however, in jaundiced, non–breast-fed infants who are more than 2 weeks old. Breast-fed infants may have elevated bilirubin during the first 3 weeks of life, but hyperbilirubinemia in these babies should be considered abnormal if the urine is dark or the stools are light. Cholestasis is diagnosed when the serum conjugated bilirubin concentration is more than 1.0 mg/dL if the total serum bilirubin concentration is less than 5.0 mg/dL or when it is more than 20% of the total serum bilirubin concentration if the total serum bilirubin concentration is less than 5.0 mg/dL.
Transient neonatal jaundice can be due to hemolysis, hepatic infection, sepsis, metabolic diseases, bile plug syndrome, or adrenal hemorrhage. Some medications and total parenteral nutrition can also cause transient jaundice. Neonatal hepatitis, biliary atresia, and choledochal malformation are the most common causes of persistent neonatal jaundice, with hepatitis and biliary atresia accounting for 70% to 80% of cases. Alagille’s syndrome and spontaneous bile duct perforation are less common causes of persistent neonatal jaundice. Caroli’s disease is a congenital abnormality of the intrahepatic bile ducts that can also cause persistent jaundice, but clinical presentation more commonly occurs in childhood or adolescence.
Biliary atresia is the most common cause of persistent neonatal jaundice that requires surgical intervention. Other less common surgically amenable causes of persistent neonatal jaundice include inspissated bile syndrome, choledochal malformation, and spontaneous perforation of the bile ducts. The initial imaging study in a child with neonatal jaundice is usually an ultrasound examination of the abdomen and pelvis. The liver size, vasculature, parenchymal echogenicity and echotexture, and intrahepatic and extrahepatic biliary ducts are evaluated with gray-scale and Doppler imaging. If a gallbladder is present, it is evaluated for size, wall thickness, presence of stones or sludge, and pericholecystic fluid. The spleen and pancreas are also evaluated. Ultrasound of the abdomen and pelvis provides good anatomic evaluation for potential sources of cholestasis; hepatocyte function and biliary drainage can be evaluated with nuclear medicine hepatobiliary scintigraphy, preferably performed with 99m Tc-mebrofenin.
Biliary Atresia
Background
Biliary atresia is a progressive, fibrosing, obliterative disease that affects the biliary tree. Biliary atresia typically begins in the neonatal period but may begin before birth. Biliary atresia results in cholestasis, which then leads to hepatic fibrosis and cirrhosis. Children with biliary atresia typically present with persistent neonatal jaundice and often also have icterus or clay-colored (acholic) stool. Biliary atresia occurs in 1 of 8000 to 16,700 live births, with a higher incidence in Asian populations.
Several classification systems for biliary atresia have been developed, generally based on which part of the biliary tree is involved and to what degree. The classification system for biliary atresia initially proposed and Ohi is one of the more widely used. In the system developed by Ohi, type I is atresia of the common bile duct, with patency of the proximal biliary ducts (right hepatic, left hepatic, and common hepatic), and it is considered to be the surgically “correctable” type of biliary atresia. Type II is atresia of the hepatic duct. Type III is involvement of the extrahepatic biliary tree and intrahepatic ducts of the porta hepatis. Varying degrees of atresia may occur in the distal ducts, ranging from hypoplasia to fibrosis, aplasia, or a miscellaneous combination of these degrees of atresia (subtypes a to d, respectively). Involvement of the proximal ducts is further divided into subtypes that are assigned lowercase Greek letters: alpha (α) is dilation of the proximal ducts, beta (β) is hypoplasia of the proximal ducts, gamma (γ) involves a bile lake at the porta hepatis, mu (µ) is fibrosis of the proximal ducts, nu (ν) is a fibrous mass at the porta hepatis, and omicron (ο) is aplasia of the proximal ducts ( Figs. 119-4 and 119-5 ).


Associated Anomalies and Malformations
Approximately 70% to 85% of infants with biliary atresia will not have any other anomalies or malformations, which has been referred to as perinatal biliary atresia. Typically, these children are born without jaundice, but jaundice develops and stools become progressively acholic within the first 2 months of life.
Approximately 10% to 15% of infants with biliary atresia have associated laterality malformations, which is known as biliary atresia splenic malformation or embryonal biliary atresia. Abnormalities associated with biliary atresia include situs inversus, symmetric bilobed liver, asplenia or polysplenia, intestinal malrotation, interrupted or absent inferior vena cava, preduodenal portal vein, aberrant hepatic artery, and cardiac anomalies. If polysplenia or situs inversus is encountered in a jaundiced infant, biliary atresia is almost invariably present. The association is important because the complex anatomy in patients with laterality malformations complicates the surgical treatment of biliary atresia.
The remaining 5% to 10% of biliary atresia cases are associated with other congenital malformations, including intestinal atresia, imperforate anus, kidney anomalies, and cardiac anomalies.
Treatment
If it is untreated, biliary atresia is fatal, with an average survival of 18 months. In patients with biliary atresia, the goal is to achieve adequate biliary drainage with the Kasai procedure (also known as the Kasai portoenterostomy, hepatic portoenterostomy, and hepatoportoenterostomy). The Kasai procedure involves excision of the obliterated biliary remnant with anastomosis of the portal plate to the small bowel with a Roux-en-Y hepatojejunostomy. For the biliary atresia to be considered surgically correctable, a portion of the proximal common hepatic duct must be patent, which can be directly anastomosed to the jejunum after the resection of the fibrotic bile duct remnant, ideally preventing the long-term sequelae of biliary atresia and the need for liver transplantation. Unfortunately, the truly correctable type of biliary atresia is uncommon, accounting for only approximately 10% to 15% of cases. Even though the remaining forms of biliary atresia are not considered correctable surgically, the Kasai procedure is used palliatively until a liver transplant is needed, with a survival rate of more than 95%. Adequate biliary drainage after a Kasai procedure has been defined as a total bilirubin concentration of less than 2.0 mg/dL anytime within the first 3 months after surgery.
It was initially accepted that the Kasai procedure should be performed before an infant diagnosed with biliary atresia reaches 2 months of age. Subsequently, however, this was not the conclusion of a more comprehensive analysis from the Japanese registry, nor was it the conclusion of Davenport, who reported a 5-year 45% transplant-free survival in a smaller cohort of children who underwent the Kasai procedure at more than 100 days of age. A collaborative North American study, which prospectively evaluated 530 patients with biliary atresia from 2004 to 2010, found that infants younger than 75 days undergoing the Kasai procedure were no more likely than infants older than 75 days to achieve adequate biliary drainage (total bilirubin concentration of less than 2.0 mg/dL within the first 3 months). In the same study, however, Superina and colleagues did show that having the Kasai procedure done before 75 days of age was associated with improved transplant-free survival, suggesting that efforts to identify and to treat infants with biliary atresia as early as possible are worthwhile.
Cholangitis is a serious potential complication of the Kasai procedure that can lead to sudden cessation of bile drainage. The greatest risk factor for development of cholangitis after a Kasai procedure is inadequate biliary drainage. Cholangitis should be suspected when the child has fever and acholic stools or fever and jaundice with an elevated direct bilirubin level and positive C-reactive protein result. Some investigators expand the diagnosis of cholangitis to include any unexplained fever in a patient who has had a Kasai procedure. Most patients who have undergone the Kasai procedure have at least one episode of cholangitis, and 90% occur in children younger than 2 years. Episodes of cholangitis in this population have been shown to adversely affect survival rates.
Imaging
Ultrasound, nuclear medicine hepatobiliary scintigraphy, CT, and MRI are the primary imaging modalities used in the work-up for biliary atresia. When necessary, transhepatic cholangiography, intraoperative cholangiography, and traditional angiography may be used. Preoperative definition of the patient’s anatomy with Doppler ultrasound, CT, MRI, and angiography is used in technical planning of the surgical procedure. Traditional angiography may be necessary to demonstrate the location and size of the portal trunk in cases in which it is not seen by CT or MRI.
Ultrasound should be the initial imaging modality for infants with cholestatic jaundice and suspected biliary atresia. The infant is kept fasted for as close to 4 hours as possible before the ultrasound examination in an attempt to maximize distention of the biliary tree and gallbladder, if one is present. A high-frequency linear transducer or microconvex transducer is used for optimal visualization of the biliary system.
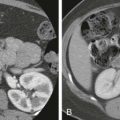
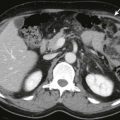
Sonographic findings in children with biliary atresia are variable. The liver parenchyma may be normal in echogenicity and echotexture, or there may be absence of identifiable bile ducts resulting in a homogeneous appearance of the liver parenchyma ( Fig. 119-6 ). If the patient presents later in the course of the disease, findings of cirrhosis and portal hypertension may be present ( Fig. 119-7 ). The common bile duct may not be seen. If the liver is not interrogated by color Doppler imaging, the hepatic artery can be mistaken for the common bile duct, resulting in a false-negative study. False-negative examination results can also occur when there is a patent portion of the common bile duct or if there is cystic dilation of the extrahepatic duct.


When the hepatic artery is correctly identified, its size may be helpful in making the diagnosis of biliary atresia. It has been shown that the diameter of the right hepatic artery in patients with biliary atresia (1.9 ± 0.4 mm) is significantly larger than the diameter of the hepatic artery in infants with hepatitis (1.4 ± 0.3 mm) and infants in the control group (1.2 ± 0.2 mm). In the same study, the hepatic artery diameter–to–portal vein diameter ratio in the biliary atresia group (0.52 ± 0.12) was larger than that in hepatitis (0.40 ± 0.07) and control (0.40 ± 0.10) groups ( P < .001). On the basis of these findings, cutoff values for diagnosis of biliary atresia were 1.5 mm (sensitivity, 92%; specificity, 87%; accuracy, 89%) for hepatic artery diameter and 0.45 for hepatic artery diameter–to–portal vein diameter ratio (sensitivity, 76%; specificity, 79%; accuracy, 78%).
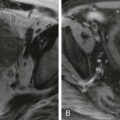
Another ultrasound finding that can be helpful in the diagnosis of biliary atresia is a focal triangular or tubular hyperechoic structure just anterior and slightly superior to the main portal vein bifurcation ( Fig. 119-8 ), known as the triangular cord sign. This echogenic structure represents the fibrotic remnant of the obliterated common bile duct. Flow is not seen within the triangular cord on Doppler evaluation. The triangular cord may not be obvious in the early stages of the disease, or it may be obscured by a large right hepatic artery. The triangular cord can also be masked by diffuse periportal hyperechogenicity due to inflammation or cirrhosis, or it may be difficult to appreciate if the fibrotic cord is small. Short-term follow-up ultrasound is warranted if the infant has findings on initial ultrasound that appear normal or that suggest neonatal hepatitis but the cholestatic jaundice is not improving or is worsening.

A triangular cord larger than 4 mm in a neonate with cholestatic jaundice has been shown to be 80% sensitive and 98% specific and to have positive and negative predictive values of 94% for the diagnosis of biliary atresia. Round, linear, or tubular hypoechoic or cystic lesions within the triangular cord have been described that are cleftlike cystic lesions within the fibrotic mass histopathologically. These cystic clefts can also be seen as triangular areas of increased signal intensity on T2-weighted MRCP images.
The gallbladder is usually small or absent in patients with biliary atresia, but visualization of a normal gallbladder does not exclude the diagnosis of biliary atresia. Jaundiced, preterm infants receiving total parenteral nutrition without biliary atresia can also have small gallbladders. The gallbladder ghost triad has been described in association with biliary atresia: an atretic gallbladder (<1.9 cm long); thinned mucosa or lack of a smooth, complete echogenic gallbladder mucosal lining with indistinct walls; and a knobby, irregular or lobular gallbladder contour ( Fig. 119-9 ). These findings are presumed to be caused by the atretic state, immaturity, and lack of function of the gallbladder in infants with biliary atresia. Gallbladder wall thickening is not a feature of biliary atresia, but it has been described in 25% of jaundiced infants without biliary atresia and may be useful in directing the work-up away from the diagnosis of biliary atresia.

When ultrasound is not diagnostic of biliary atresia, the next diagnostic test is usually a nuclear medicine hepatobiliary scintigraphy study. This study is performed with 99m Tc and an iminodiacetic acid derivative, preferably mebrofenin, because of its higher hepatic excretion. Ideally, a 3- to 7-day course of oral phenobarbital (5 mg/kg/day in two divided doses) is given to the child in preparation for the examination because it is thought that phenobarbital enhances bile excretion and flow, which can help differentiate biliary atresia from neonatal hepatitis. If the infant is approaching the age before which a Kasai procedure is ideally performed (60-75 days), the hepatobiliary study may be attempted without the phenobarbital preparation or with a shorter 2- to 3-day pretreatment with ursodeoxycholic acid (20 mg/kg/day in two divided doses, continued until the test is finished) in an effort to save time and to prevent a delay in the diagnosis.
According to the practice guidelines of the Society of Nuclear Medicine, an infant who is having a hepatobiliary scan should be kept fasted for at least 2 hours before the intravenous administration of 1 mCi of 99m Tc-mebrofenin. Anterior images are then acquired dynamically, starting at the time of injection and continuing for 60 minutes, at 1 minute per frame. Delayed images in the anterior and right lateral projections are obtained after a diaper change, including 5-minute images at 4 to 6 hours and 10-minute images at 24 hours if no radiopharmaceutical has been visualized in the small bowel.
The findings on the hepatobiliary scintigraphy study are considered normal if the hepatic parenchyma is visualized almost immediately after intravenous administration of the isotope, followed sequentially by activity in the intrahepatic biliary ducts, the extrahepatic biliary ducts, the gallbladder, and the small bowel, ideally within 1 hour after administration of the isotope. Neonatal hepatitis is presumed when there is delayed hepatocyte clearance of the radiopharmaceutical but activity is seen in the small bowel by 24 hours. If the radiopharmaceutical is excreted into the bowel by 24 hours, the diagnosis of biliary atresia is essentially excluded. When no radiopharmaceutical is seen in the bowel on the 24-hour delayed images, biliary atresia should be strongly considered ( Fig. 119-10 ). However, nonvisualization of the radiopharmaceutical in the bowel at 24 hours is not highly specific for biliary atresia because hepatocellular dysfunction in infants with neonatal hepatitis can sometimes be pronounced enough that the radiopharmaceutical is not excreted within the first 24 hours. The reported diagnostic accuracy of hepatobiliary scintigraphy in the diagnosis of biliary atresia is 56% to 81.6%, with a sensitivity of 91.7% to 100% and a specificity of 35% to 76.9%.

For biliary atresia to be excluded, the patency of the common hepatic duct and common duct should be demonstrated, but this is not always achieved with ultrasound and hepatobiliary scintigraphy. MRCP may eliminate the need in children for more invasive imaging, such as percutaneous transhepatic or intraoperative cholangiography, biopsy, or exploratory surgery, if normal biliary tract anatomy can be demonstrated. The diagnosis of biliary atresia is made on MRCP when the common bile duct or the common hepatic duct is not visualized. In infants with cholestatic jaundice, Han and colleagues demonstrated that MRCP had an accuracy of 98%, sensitivity of 100%, and specificity of 96% for the diagnosis of biliary atresia. As on ultrasound, MRCP findings of biliary atresia include nonvisualization of the extrahepatic bile ducts, a small or absent gallbladder, and periportal fibrosis (the triangular cord sign on ultrasound). Periportal fibrosis is often seen as a triangular area of high signal intensity ventral to the porta hepatis on the T2-weighted images and as a hypointense signal paralleling the portal vein branches on T1-weighted sequences that becomes isointense with liver after administration of gadolinium. Gadolinium administration distinguishes the enhancing periportal fibrosis from the nonenhancing, dilated periportal ducts seen in other disease processes. On T2-weighted images, a focal region of hyperintense signal thought to be fluid has been described within the high signal intensity periportal fibrosis in some children with biliary atresia ( Fig. 119-11 ).

False-positive MRCP results for biliary atresia have been reported; these are in part due to the fact that MRCP relies on the production and excretion of bile into the bile ducts to visualize the biliary tract. Children with disease states that result in decreased hepatobiliary function and subsequent decreased bile production, such as sclerosing cholangitis, may have false-positive examination findings because the biliary tree is not visualized on MRCP owing to a relative lack of bile within the ducts.
Liver biopsy can help confirm the diagnosis of biliary atresia. An 18-gauge needle biopsy of the liver has been shown to be 90% sensitive, 96% specific, and 85% to 93% accurate for the diagnosis of biliary atresia in cholestatic children. Neonatal hepatitis and early biliary atresia may have identical ultrastructural and light microscopic features. If the biopsy is performed early in a particular patient’s disease process, the typical ultrastructural features of biliary atresia (i.e., proliferation of the intralobular bile ductules with loss of ductular microvilli, periductal inflammatory fibrosis, and bile plugging) may not yet be present, and a false-negative result may occur. If the clinical course and liver biopsy results are not concordant, repeated biopsy may be warranted.
In some children, ultrasound, nuclear scintigraphy, MRCP, and liver biopsy results may all be inconclusive. In these children, visualization of the bile ducts by cholangiography intraoperatively, percutaneously through the liver or gallbladder, or by endoscopy may be necessary.
In children with biliary atresia who have undergone the Kasai procedure, it is important to monitor for changes of fibrosis, cirrhosis, and portal hypertension on postoperative imaging. It is also important to know the patient’s baseline anatomy and to remember that the loop of jejunum that is anastomosed to the porta hepatis can be seen as a fluid-filled structure, which can be mistaken for a postoperative fluid collection. If necessary, patency of the anastomosis can be confirmed by nuclear scintigraphy.
Alagille’s Syndrome
Background
Alagille’s syndrome, also known as arteriohepatic dysplasia, is a rare autosomal dominant disease characterized by chronic cholestasis due to a paucity of interlobular bile ducts relative to the number of portal areas within the liver. Children with Alagille’s syndrome usually present as neonates but may present later. For diagnosis, at least three of the five typical features of the disease are exhibited: chronic cholestasis from interlobular bile duct paucity, congenital heart disease typically involving hypoplasia or stenosis of the peripheral pulmonary arteries, “butterfly” vertebrae, posterior embryotoxon (an ocular abnormality), and peculiar facies.
Pulmonary artery stenosis at various levels is one of the most common manifestations of Alagille’s syndrome. Other associated vascular anomalies include basilar or middle cerebral artery aneurysms, internal carotid artery anomalies, aortic coarctation or aneurysm, and moyamoya syndrome. The intracranial vascular anomalies account for up to 34% of the mortality in these patients. Renal artery involvement may result in hypertension.
Imaging
The sonographic findings of Alagille’s syndrome are similar to those of neonatal hepatitis. In neonatal hepatitis, the liver size and echogenicity may be normal or increased, and the intrahepatic bile ducts are typically not visualized. Nuclear hepatobiliary scintigraphy may initially resemble biliary atresia and typically fails to show normal excretion of the radiopharmaceutical into the bowel ( Fig. 119-12 ). Cholangiography or MRCP should demonstrate patency of the extrahepatic bile ducts, excluding the diagnosis of biliary atresia, but because Alagille’s syndrome is rare, this diagnostic step is occasionally foregone, leading to treatment with the Kasai procedure before the pathologic diagnosis of Alagille’s syndrome is made.