Chapter Outline
Imaging Approach for a Suspected Hepatic Mass
Benign Hepatic Neoplasms and Neoplasm-Like Abnormalities
Focal Inflammatory-Infectious Lesions of the Liver
Cirrhosis and Diffuse Disorders of the Liver
Liver disease is less common in the pediatric population than it is in the adult population, but the sequelae can be equally devastating and may lead to the need for liver transplantation. This chapter addresses the more common congenital, neoplastic, infectious, vascular, and metabolic disorders of the pediatric liver.
At birth, the liver occupies approximately 40% of the abdominal cavity and represents 5% of the infant’s total body weight. The relative size of the liver to the abdominal cavity decreases as a child grows. During infancy, the inferior hepatic margin is normally several centimeters below the costal margin, but it becomes less prominent throughout childhood.
Imaging of the pediatric liver may be prompted by hepatomegaly, abnormal liver function test results, jaundice, a palpable mass, or an abnormality detected on prenatal ultrasound. Imaging also plays an important role in evaluation of the liver before and after liver transplantation. This chapter focuses on liver masses and diseases that are unique to the pediatric population, particularly when the imaging approach diverges from that used in the adult population.
Imaging Approach for a Suspected Hepatic Mass
A palpable abdominal mass is the most common clinical presentation in a child with a liver mass. Other frequently encountered presenting signs and symptoms include hepatomegaly, abdominal pain, weight loss, anorexia, jaundice, and paraneoplastic syndromes. In addition, large vascular malformations may be manifested as congestive heart failure or consumptive coagulopathy (Kasabach-Merritt syndrome). Ultrasound is a useful imaging modality in the work-up of a suspected liver mass in a child because it allows evaluation of the abdominal viscera and vasculature. Ultrasound is relatively inexpensive, does not expose the child to ionizing radiation, and usually does not require that the patient be sedated. When an abdominal mass is identified, ultrasound can help determine the organ of origin, whether the mass is cystic or solid, if there is vascular flow to the mass, whether the mass contains calcifications, and involvement of other abdominal organs.
After the presence of a liver mass is confirmed by ultrasound, additional imaging with computed tomography (CT) or magnetic resonance imaging (MRI) is usually performed. Because surgical resection is the first line of treatment for many pediatric liver masses, precise anatomic delineation of the mass is important. The decision to pursue CT or MRI as the subsequent imaging modality is often driven by institutional and individual preferences because each modality has its own strengths and limitations. MRI is often preferred because it provides detailed multiplanar anatomic information, does not rely on proper timing of bolus contrast material administration for delineation of the vasculature, and imparts no ionizing radiation. CT is often more readily available than MRI and less commonly requires sedation.
When focal nodular hyperplasia is a consideration, technetium Tc 99m sulfur colloid scintigraphy may be a helpful adjunct study. Focal nodular hyperplasia demonstrates normal to increased uptake of the radiopharmaceutical, whereas other hepatic masses create photopenic areas. Tagged red blood cell studies can also be helpful in differentiating hemangiomas from other masses in the liver; hemangiomas demonstrate increased radiopharmaceutical activity, whereas other hepatic masses do not.
Congenital Anomalies
Malformations and Hernias
The major congenital malformations of the liver include anomalous lobar development, lobar hypoplasia, and complete lobar agenesis. Agenesis and hypoplasia of the liver most commonly involve segment IV in the left lobe and segments V and VIII in the right lobe. The best diagnostic clue, besides obvious absence of liver parenchyma, is absence of the respective hepatic and portal veins and intrahepatic biliary ducts. With congenital agenesis or hypoplasia of a lobe of the liver, compensatory enlargement of the remaining hepatic parenchyma usually occurs. Hepatic agenesis is associated with biliary tract disease, portal hypertension, and other congenital anomalies. Herniation of the liver parenchyma through a diaphragmatic hernia is typically present at birth but may be delayed. For example, group B streptococcal pneumonia has been associated with late-onset, right-sided diaphragmatic hernia.
Fibropolycystic Diseases of the Liver
Fibropolycystic diseases of the liver are thought to be the sequelae of ductal plate malformations, which involve partial or total arrest of remodeling of the ductal plate and result in persistence of embryonic biliary structures. Ductal plate malformations may involve segmental bile ducts (large), interlobular bile ducts (medium sized), or the smallest bile duct ramifications, resulting in distinct disease entities. Choledochal malformations (extrahepatic) and Caroli’s disease (intrahepatic) involve malformations of large embryologic hepatobiliary ducts. Autosomal dominant polycystic liver disease (ADPLD) involves malformations of medium-sized embryologic hepatobiliary ducts. Fibropolycystic liver diseases involving malformations of small embryologic hepatobiliary ducts include congenital hepatic fibrosis and biliary hamartomas (von Meyenburg complexes). Caroli’s syndrome (Caroli’s disease with congenital hepatic fibrosis) involves large and small biliary ducts.
Choledochal malformations (also known as choledochal cysts) and Caroli’s disease (also known as type V choledochal cyst) are characterized by dilated extrahepatic and intrahepatic bile ducts, respectively, and are thought to happen during early bile duct embryogenesis. A second widely accepted theory of development of choledochal malformations is abnormal proximal insertion of the pancreatic duct into the common bile duct, which causes reflux of pancreatic enzymes into the common bile duct that weaken the duct wall and results in abnormal dilation. Caroli’s disease and choledochal malformations, their diagnostic work-up, and imaging findings are discussed in Chapter 119 .
ADPLD involves malformations of medium-sized bile ducts and is associated with autosomal dominant polycystic kidney disease (ADPKD) ( Fig. 120-1 ). ADPLD is very rarely seen without ADPKD. The development of liver cysts usually occurs after the development of renal cysts, and because renal cysts in ADPKD typically are not manifested until the second or third decade of life, ADPLD rarely is manifested in childhood. ADPLD is manifested in 29% to 48% of people with ADPKD. Hepatomegaly is usually the first indication that there may be underlying polycystic liver disease. Ultrasound, CT, MRI, and magnetic resonance cholangiopancreatography (MRCP) are useful in the detection of cysts in the liver. Ultrasound demonstrates multiple anechoic, thin-walled lesions, with posterior acoustic enhancement. On CT, the hepatic cysts are fluid attenuation; on MRI and MRCP, the cysts are hypointense on T1-weighted sequences and hyperintense on T2-weighted sequences. If there has been hemorrhage into the cysts, prior infection, or other reasons that the fluid may be more complex, the echogenicity, attenuation, and signal will change accordingly.

Congenital hepatic fibrosis involves defective remodeling of the smaller interlobular bile ducts and is characterized by abnormal branching of the intrahepatic portal veins and progressive fibrosis of the portal tracts. Congenital hepatic fibrosis may occur with or without macroscopically visible cystic dilations of the intrahepatic biliary ducts. Congenital hepatic fibrosis with macroscopic liver cysts that communicate with the biliary ducts (also known as Caroli’s disease) is referred to as Caroli’s syndrome. Caroli’s disease is much rarer than Caroli’s syndrome. Caroli’s disease and Caroli’s syndrome can occur in different members of the same family, suggesting that these two disease entities may be on the same continuum. Congenital hepatic fibrosis sometimes is manifested in early childhood but may not be manifested until later in life.
Congenital hepatic fibrosis and Caroli’s syndrome are associated with fibrocystic renal disease, including autosomal recessive polycystic kidney disease (ARPKD) and ADPKD; glomerulocystic kidney disease; diffuse cystic dysplastic kidneys; tubulointerstitial disorders, such as nephronophthisis, chronic tubulointerstitial disease, and urine-concentrating defect; and medullary sponge kidney (a radiologic term for microcystic dilations of renal collecting ducts on contrast-enhanced imaging that may be seen with or without nephrocalcinosis). All patients with ARPKD who survive the neonatal period develop clinical findings of congenital hepatic fibrosis. Nonobstructive dilation of the intrahepatic bile ducts in the liver (Caroli’s disease) is seen at the histologic level in a subset of patients as well.
On ultrasound, patients with congenital hepatic fibrosis may have normal or increased parenchymal echogenicity, normal or coarsened parenchymal echotexture, hyperechoic portal triads, or poorly defined portal vessels. Cysts may be seen in the parenchyma, and the left lobe of the liver may be enlarged; there may be signs of portal hypertension, including reversal of flow in the portal vein, splenomegaly, and varices. Because of the association with fibrocystic kidney disease, the kidneys may be abnormal as well, and evaluation with a high-frequency transducer may be useful for detailed evaluation of the renal parenchyma. On CT, the liver is heterogeneous in attenuation, the left lobe of the liver is enlarged, and the associated findings of portal hypertension and fibrocystic renal disease may be evident. Findings of congenital hepatic fibrosis on MRI include hypointense signal in the liver parenchyma on T1- and T2-weighted sequences with hyperintense signal within the portal vessels on T2-weighted imaging (which reflects the periportal fibrosis), enlarged spleen, and accompanying fibrocystic changes in the kidneys. When associated Caroli’s disease is present, MRCP may reveal cystic or fusiform dilations and irregularities of the intrahepatic bile ducts. Complications of congenital hepatic fibrosis include ascending cholangitis, sequelae of portal hypertension including cirrhosis and variceal bleeding, and increased risk of hepatocellular carcinoma.
Biliary hamartomas (also known as biliary microhamartomas or von Meyenburg complexes) are also thought to be due to small interlobular ductal plate malformations. Biliary hamartomas can be confused with metastatic disease, and therefore definite diagnosis requires a tissue sample. On ultrasound, biliary hamartomas may be hypoechoic or hyperechoic. On CT, biliary hamartomas typically are uniform in size and do not enhance. On MRI, biliary hamartomas are typically hypointense on T1-weighted sequences and hyperintense on T2-weighted sequences.
Hepatic Cysts
Hepatic cysts are thought to represent growth arrest and dilation of the biliary tract, although there is some support for a theory of vascular insult leading to necrosis and cyst formation ( Fig. 120-2 ). They may be detected prenatally, but a definitive diagnosis may not be possible until after birth. Differential diagnosis for peripheral lesions includes cystic hepatoblastoma, mesenchymal hamartoma, and vascular malformation. When the cyst has a subhepatic location, choledochal cyst, pancreatic pseudocyst, alimentary duplication, mesenteric cyst, and urachal cyst are additional considerations.

Malignant Hepatic Neoplasms
In children, hepatic masses are relatively uncommon, accounting for 5% to 6% of all abdominal masses in the pediatric population. Primary neoplasms of the liver represent only 0.5% to 2% of all childhood malignant neoplasms, but they are by far the most common primary malignant neoplasms of the pediatric gastrointestinal tract. Primary hepatic neoplasms are the third most common abdominal malignant neoplasm in childhood, after Wilms’ tumor and neuroblastoma.
Primary Malignant Neoplasms
No hepatic malignant neoplasm has imaging features so pathognomonic as to obviate tissue diagnosis. The primary purpose of imaging is to characterize the lesion and to define the extent of disease relative to segmental anatomy, vascular structures, and the biliary system for potential chemotherapeutic intervention and preoperative planning. Follow-up imaging is necessary for assessment of tumor response to treatment protocols and determination of the potential for resection. Most malignant pediatric liver tumors require complete resection or transplantation as a prerequisite for cure.
Hepatoblastoma, hepatocellular carcinoma, and the less frequently encountered embryonal sarcoma account for approximately two thirds of primary malignant hepatic tumors in children. The remaining primary tumors of the liver are uncommon in children and include fibrolamellar hepatocellular carcinoma, angiosarcoma, epithelioid hemangioendothelioma (a very rare tumor that is on the continuum between angiosarcoma and hemangioma), rhabdoid tumor, endodermal sinus tumor, and lymphosarcoma. Embryonal rhabdomyosarcoma is the most common malignant neoplasm to arise from the biliary tract in children and is discussed in Chapter 119 .
Hepatoblastoma
Hepatoblastoma is the most common pediatric hepatic malignant neoplasm, accounting for 1% of all pediatric malignant neoplasms. Hepatoblastomas are embryonal tumors derived from pluripotent stem cells with the capability of differentiating into hepatocytes and biliary epithelial cells. On histologic examination, these tumors are classified as epithelial, mixed (i.e., epithelial and mesenchymal), or anaplastic type. The epithelial type is the most common, accounting for approximately 60% of all hepatoblastomas. The epithelial type is further subdivided into fetal and embryonal forms. Pure fetal histology carries a better prognosis, and anaplastic or undifferentiated histology carries a poorer prognosis. With the mixed-type tumors, the presence of mesenchymal components carries a more favorable prognosis. The most commonly found mesenchymal components are cartilage and osteoid.
Hepatoblastoma usually occurs in infants and children younger than 3 years, with a median age of occurrence of approximately 1 year. This tumor is slightly more common in boys than in girls, with a male-to-female ratio of approximately 3 : 2. No environmental risk factors have been definitively linked to hepatoblastoma, but an increased incidence of hepatoblastoma has been reported in infants with prematurity or low birth weight. Hepatoblastomas occurring within families have also been reported, with increased risk in children with Beckwith-Wiedemann syndrome and familial adenomatous polyposis. The association between hepatoblastoma and these two syndromes suggests that aberrations of chromosomes 11 and 5 play a role in pathogenesis. Several other conditions that demonstrate an increased incidence of hepatoblastoma are hemihypertrophy, Wilms’ tumor, and biliary atresia.
Hepatoblastoma usually is manifested as a painless abdominal mass, but weight loss, anorexia, abdominal pain, jaundice, and rarely precocious puberty may be present. Most patients have no underlying liver disease. On occasion, levels of human β-chorionic gonadotropin are elevated, producing virilization and moderate thrombocytosis. Serum α-fetoprotein levels are elevated in more than 90% of children with hepatoblastoma. In a child younger than 3 years with a liver mass and an elevated α-fetoprotein level, hepatoblastoma should be the primary consideration.
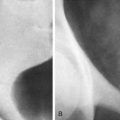
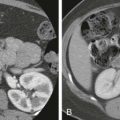
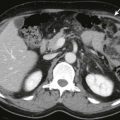
The most common site of involvement of hepatoblastoma is the right lobe of the liver, although bilateral disease is seen in approximately 35% of cases ( Figs. 120-3 and 120-4 ). Distant metastases are present in 20% of patients at the time of diagnosis, most commonly within the lungs. On pathologic examination, hepatoblastomas have a tendency to be pseudoencapsulated, well defined, and large at the time of presentation. On average, these liver masses are 10 to 20 cm in diameter at presentation. Hepatoblastoma commonly appears sonographically as a well-circumscribed, predominantly echogenic, solid mass. Calcifications are apparent by ultrasound in up to one third of patients and are manifested as punctuate heterogeneous hyperechoic areas with posterior shadowing. Rarely, hepatoblastomas are cystic.


CT usually demonstrates a large, hypoattenuating, well-defined mass with homogeneous enhancement after the intravenous administration of contrast material ( Fig. 120-5 ). CT better defines calcifications than ultrasound or MRI. Calcifications tend to be punctate and delicate in epithelial-type tumors, but they are more often extensive and coarse in the mixed-type tumors. MRI most commonly depicts a well-defined hepatic mass that is hypointense on T1-weighted sequences and hyperintense on T2-weighted sequences. When necrosis or hemorrhage exists within the tumor, there may be regions of hyperintense T1-weighted signal within the mass. Contrast-enhanced CT and MRI readily depict invasion, encasement, or thrombosis of the main portal vein or the hepatic artery. The presence or absence of these findings strongly affects tumor resectability. The advent of combined adjuvant and neoadjuvant chemotherapy, hepatic resection, and, in some cases, liver transplantation has led to a dramatic improvement in survival. The 5-year survival rate for children with hepatoblastoma is 75%.

Hepatocellular Carcinoma
Hepatocellular carcinoma is rare in childhood, accounting for approximately 0.5% of all pediatric malignant neoplasms. After hepatoblastoma, hepatocellular carcinoma is the second most common primary hepatic tumor of childhood and the most common primary liver tumor in children older than 4 years. Histologic features of hepatic cellular carcinoma in children are similar to those in adults, with tumor cells that closely resemble hepatocytes. Tumor growth varies, but these lesions often are manifested as multicentric, diffuse, or infiltrative masses. If necrosis is present, portions of the tumor may appear cystic. Hepatocellular carcinoma occurs more commonly in boys and older children. It has a bimodal age distribution. The first peak occurs at age 4 to 5 years, and the second peak occurs at age 12 to 14 years. The mean age at diagnosis is 12 years.
Although histologic and gross specimen characteristics follow the patterns seen in adults, the underlying causes in children are for the most part unique: biliary atresia, Fanconi anemia, hepatitis B viral infection, familial cholestasis, hereditary tyrosinemia, and glycogen storage disease type 1A. Glycogen storage disease type 1A results from a deficiency of glucose-6-phosphatase and leads to the development of hepatic adenomas in approximately 50% of patients. These patients are also at risk for hepatocellular carcinoma from malignant transformation of existing adenomas or from development of new tumors. Hyperalimentation, treatment with androgenic steroids or methotrexate, and use of oral contraceptives are also associated with an increased risk for hepatocellular carcinoma. More than 50% of children with hepatocellular carcinoma have no preexisting liver disease or other risk factors, however. Many pediatric hepatocellular carcinomas are de novo cases that usually are not related to hepatic cirrhosis or other preexisting liver disease. This is in contrast to hepatocellular carcinoma in adults, in whom 70% to 90% of cases are related to cirrhosis.
Despite differences in etiology, the macroscopic and microscopic features of hepatocellular carcinoma in children and adults are similar. The classic trabecular pattern and fibrolamellar variants are the two most common types of hepatocellular carcinoma in children. It has been observed that there are improved outcomes associated with the fibrolamellar subtype in adults, but this has not been reported in the pediatric population.
Hepatocellular carcinoma frequently is manifested with abdominal fullness associated with pain or discomfort. Other common signs and symptoms include weight loss and fatigue. The most common clinical finding on physical examination is hepatomegaly. At presentation, between 60% and 80% of children with hepatocellular carcinoma have elevated serum α-fetoprotein levels. Metastatic disease at the time of presentation is common, and typical sites of metastasis include regional lymph nodes, lungs, and bone. Therapeutic results are uniformly poor, with an overall survival rate of about 20%. Complete tumor excision remains the only chance for long-term survival. Because of a high rate of metastatic disease at presentation (up to 50% of children) and because hepatocellular carcinoma in children is often multifocal, complete tumor excision is possible in only a minority of cases. The question of optimal therapy for pediatric hepatocellular carcinoma remains controversial, and the role of liver transplantation remains a matter for debate.
The imaging findings for hepatocellular carcinoma in children do not differ significantly from those seen in adults and are nonspecific, with variable appearance of the tumors on ultrasound, CT, and MRI. On ultrasound, hepatocellular carcinoma is typically hypoechoic to isoechoic compared with normal hepatic parenchyma. When the tumor is infiltrative, there may be subtle disruption of the normal liver echotexture. CT can demonstrate a solitary mass, infiltrative tumor, or multiple, confluent masses that are low attenuating to isoattenuating relative to normal hepatic parenchyma on noncontrast or portal venous phase studies. With contrast-enhanced CT, these tumors may be hyperattenuating ( Fig. 120-6 ). Calcifications are uncommon, but hemorrhage and necrosis are frequently seen. With T1-weighted MRI, hepatocellular carcinoma is frequently hypointense to normal hepatic parenchyma, but tumors may also appear isointense to hyperintense. Lesions of mixed signal intensity are seen in the setting of hemorrhage, necrosis, or fatty metaplasia. On T2-weighted images, most lesions demonstrate moderately increased signal intensity.

Undifferentiated Embryonal Sarcoma
Undifferentiated embryonal sarcoma is a rare, highly malignant hepatic neoplasm of mesenchymal origin that occurs almost exclusively in the pediatric population. It is the third most common primary hepatic malignant neoplasm of childhood. The age distribution for undifferentiated embryonal sarcoma falls between the peak ages of the incidence for hepatoblastoma and hepatocellular carcinoma, in children between the ages of 6 and 10 years, with no gender predilection. On histologic examination, this tumor consists of undifferentiated spindle cells that resemble embryonal cells with a myxoid stroma. The tumor typically is well demarcated by a fibrous pseudocapsule.
Undifferentiated embryonal sarcoma usually is manifested as a large abdominal mass, although pain, fever, jaundice, and weight loss have been reported. On occasion, children with this tumor present with an acute abdomen after the tumor ruptures. There are no reliable serum markers for this tumor, although leukocytosis and anemia may be present. Serum α-fetoprotein levels are normal.
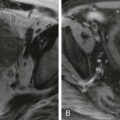
Undifferentiated embryonal sarcoma varies in sonographic appearance from a septate, cystic mass to a heterogeneous, predominantly solid lesion. The solid lesions may demonstrate high-level echoes with acoustic shadowing from calcifications. On noncontrast CT, undifferentiated embryonal sarcoma is frequently hypoattenuating. There may be enhancement of the internal septa and fibrous capsule after administration of contrast material. On MRI, undifferentiated embryonal sarcomas typically appear heterogeneous; these tumors usually are hypointense on T1-weighted sequences and hyperintense on T2-weighted sequences, corresponding to the cystic components of the mass. The fibrous pseudocapsule and internal septations appear hypointense on T1-weighted and T2-weighted sequences ( Fig. 120-7 ).

Hepatic Angiosarcoma
Hepatic angiosarcoma is a rare, high-grade malignant neoplasm of endothelial cells. Unlike hepatic angiosarcoma in the adult population, pediatric hepatic angiosarcoma affects girls more than boys, with a female-to-male ratio of approximately 2 : 1. There is a lack of clear association with environmental carcinogens such as arsenic and polyvinyl chloride. The mean age at presentation is 40 months. Pediatric hepatic angiosarcoma usually is manifested as an abdominal mass that may be associated with jaundice, abdominal pain, vomiting, fever, tachypnea, dyspnea, or anemia ( Fig. 120-8 ). Consumption coagulopathy, which frequently occurs in adults with hepatic angiosarcoma, has not been described in children.

Pediatric hepatic angiosarcoma differs histologically from adult hepatic angiosarcoma. Pediatric hepatic angiosarcoma can have hypercellular whorls of sarcomatous cells or kaposiform spindle cells similar to kaposiform hemangioendothelioma of soft tissue in addition to the features of adult angiosarcoma. This tumor typically is manifested as a rapidly enlarging abdominal mass, and it usually is unresectable at presentation. Metastatic disease, particularly to the lungs, is a common early occurrence. Pediatric hepatic angiosarcoma has been described in the background of infantile hemangioendothelioma, and it therefore may be missed on the initial biopsy specimen. Because of the possibility of an association between infantile hemangioendothelioma and pediatric hepatic angiosarcoma, a presumptive diagnosis of infantile hemangioendothelioma in a child older than 1 year should probably be followed by a biopsy. There are no clear radiographic features to distinguish the two tumors.
Epithelioid Hemangioendothelioma
Epithelioid hemangioendothelioma is a very rare malignant neoplasm of vascular origin arising in soft tissues, lung, and liver that has malignant potential intermediate between infantile hemangioendothelioma and hepatic angiosarcoma. On histopathologic examination, the tumor consists of epithelioid and dendritic cells. The center of the tumor is predominantly fibrous stroma. This tumor may infiltrate the hepatic sinusoids and portal branches and, in doing so, obliterate them. These features may be misinterpreted as veno-occlusive disease. Splenomegaly, ascites, and portal hypertension may be present. Epithelioid hemangioendothelioma typically occurs after the second decade of life and is manifested with various clinical symptoms.
Epithelioid hemangioendotheliomas tend to be multifocal or multicentric at presentation. Metastatic disease is seen at presentation in approximately 27% to 45% of patients. Ultrasound characteristics of this tumor vary and are nonspecific. Tumors have been described as hypoechoic, isoechoic, and hyperechoic. Internal calcifications may be present. The lesions are hypoattenuating relative to normal hepatic parenchyma on unenhanced CT. After intravenous administration of contrast material, there is typically peripheral enhancement followed by relatively uniform enhancement that is isoattenuating compared with hepatic parenchyma. MRI may depict tumor characteristics better than CT. On T1-weighted sequences, the tumor typically demonstrates decreased signal relative to normal hepatic parenchyma and demonstrates peripheral enhancement after intravenous administration of gadolinium. The lesions may be relatively heterogeneous on T2-weighted sequences.
Epithelioid hemangioendothelioma tumors are notable for resistance to chemotherapy and radiation therapy. The primary treatment of choice is radical hepatic resection or liver transplantation.
Secondary Malignant Neoplasms
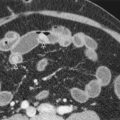
The most commonly encountered secondary or metastatic hepatic malignant neoplasms in children include neuroblastoma, Wilms’ tumor, lymphoma, and leukemia. Metastatic disease typically appears as solitary or multiple discrete masses within the hepatic parenchyma, although leukemia may be manifested with a diffuse infiltrative pattern. Stage IVS neuroblastoma, in which infants have metastases confined to the liver, skin, or bone marrow, usually is manifested as diffuse infiltration of the hepatic parenchyma. Hepatic metastases usually appear hypoechoic on ultrasound, but diffuse involvement of the liver may be difficult to detect. On CT, metastatic disease typically appears as hypoattenuating lesions, although care should be taken during portal venous phase scanning because some metastases become isoattenuating during this phase ( Figs. 120-9 and 120-10 ). Metastatic disease in the liver typically has low signal intensity on T1-weighted MR sequences and high signal intensity on T2-weighted sequences.