Clinical Presentation
The patient is a 42-year-old male with a chief complaint of left leg pain for approximately 1 year. One year ago, he developed increasing left radicular type pain in the S1 distribution with radiation to the posterior aspect of the thigh and leg to the ankle. This became more frequent. He had a magnetic resonance image (MRI) performed 10 months ago that demonstrated a soft tissue mass in an enlarged S1 lateral recess. The possibility of a schwannoma or neuroma was raised. The patient underwent conservative treatment with painkillers and had a repeat MRI scan along with a computed tomography (CT) myelogram 3 months later that showed the mass to be unchanged. He then developed increasing symptoms with numbness in his left foot.
Imaging Presentation
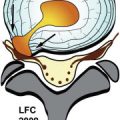
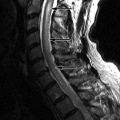
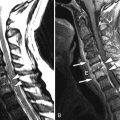
Sagittal and axial T1-weighted images demonstrate a large intermediate signal intensity soft tissue mass in an enlarged left S1 lateral recess displacing the thecal sac to the right. The L5-S1 disc is of normal height, which would not be expected if this were a massive disc herniation. This was found to be a Ewing’s sarcoma at surgery ( Fig. 32-1 ) .

Discussion
Ewing’s sarcoma (ES) is a malignant tumor that occurs mainly in children and adolescents. Ewing’s sarcoma can be divided into skeletal Ewing’s with its origin in bone and extraskeletal Ewing’s. Extraskeletal Ewing’s sarcoma has been reported to arise from numerous sites including the scalp, larynx, nasal fossa, neck, lung, extremities, paravertebral soft tissues, and the epidural space. Chromosomal and structural evaluation of both types of Ewing’s sarcoma strongly suggests that they are identical tumor types. ES is part of a group of blue, round cell tumors that include primitive neuroectodermal tumor (PNET), lymphoma, Langerhans cell granulomatosis, and neuroblastoma. It is believed that ES and PNET share a common origin from a precursor neural cell.
Skeletal ES is the second most common cancer of bone in children and adolescents and has an incidence of 2.1 per million children in the United States. Involvement of the vertebrae with ES is usually the result of metastatic disease. Primary ES of the vertebrae is much rarer, comprising 3.5% to 10% of all cases of osseous ES. Skeletal ES manifests between the ages of 5 and 30 years of age in 90% of cases, with its peak incidence in the second decade of life. ES is uncommon in children of African or Asian descent. ES has a male predominance of approximately 1.5:1. Extraskeletal ES manifests at a slightly older age (average 20 years) and occurs equally in both sexes. Unlike osteosarcoma, ES does not appear to be caused by exposure to radiation. ES most commonly occurs in the sacrum, slightly less commonly in the lumbar and thoracic regions, and least commonly in the cervical spine. Most lesions occur in the vertebral body, although lesions can extend into the posterior elements.
The most common manifesting symptom in patients with spinal ES is pain. The pain can be local or radiate to one or both extremities. A common misdiagnosis is lumbar disc disease; however, unremitting pain and/or pain at night should raise the suspicion for tumor. At initial presentation, 58% to 80% of patients will present with a neurologic deficit or radiculopathy. Cord compression is common. Other symptoms include swelling or a palpable mass and systemic symptoms including fever, which may mistakenly lead to a diagnosis of underlying infection. Approximately 20% of patients present with gross metastatic disease, but there is a high incidence of micrometastases. Because ES metastasizes to bone and lung, evaluation for metastatic disease should include nuclear medicine technetium bone scan and CT of the chest. Serum lactic dehydrogenase is the only reliable marker of tumor burden and should be closely monitored.
Imaging Features
Extraskeletal Ewing’s Sarcoma
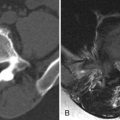
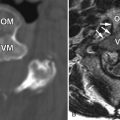
Extraskeletal ES of the spine arises in the epidural space. Plain radiographs may be of assistance only when one sees smooth enlargement of a neural foramen. CT will demonstrate a mass of intermediate density in the epidural space. Depending on the location of the mass, the neural foramen may be enlarged with smooth borders. One may see the effect of the mass on the thecal sac with myelography, plain CT, or post-myelogram CT ( Fig. 32-2 ) . On MRI, the tumor is typically low to intermediate T1 signal intensity, high T2 signal intensity, and exhibits heterogeneous enhancement ( Fig. 32-3 ) .


Skeletal Ewing’s Sarcoma
Plain radiographs demonstrate imaging findings late in skeletal ES, often after neurologic signs have appeared. The most common finding in skeletal ES is lytic bone destruction of the vertebrae, which can vary from the more common focal tiny perforation-like lytic lesions to vertebra plana. A small proportion of ES may show reactive sclerotic change. CT can demonstrate the extent of bone involvement and any soft tissue component. On CT, ES appears as a permeative intramedullary mass with a possible extraosseous soft tissue mass ( Fig. 32-4 ) . There may be heterogeneous enhancement of both the intraosseous and extraosseous components of the mass and also areas of central necrosis. CT can be important diagnostically to separate ES from osteogenic sarcoma because the latter often has a matrix of bone that can be seen on CT and occasionally on plain radiographs. Magnetic resonance imaging (MRI) is superior to CT in demonstrating the relationship of the tumor to the spinal canal and its contents, the adjacent vasculature, and the extent of bone marrow abnormality. The tumor exhibits signal abnormality that is typical of most malignant tumors: intermediate signal intensity on T1-weighted images and intermediate to high T2-weighted signal intensity. The tumor demonstrates moderate enhancement that may appear homogeneous or heterogeneous depending on intrinsic regions of tumor necrosis ( Fig. 32-5 ) .