Abstract
Fryns syndrome is a rare autosomal recessive disorder characterized by diaphragmatic defects, distal limb hypoplasia, facial dysmorphism, and associated major anomalies. It is the most common syndrome associated with congenital diaphragmatic hernia. The pathogenesis of Fryns syndrome is not well understood, but abnormal neural crest cell migration may play a role. Although genetic loci for this disorder are currently under investigation, diagnosis is solely based on clinical criteria. Multiple other chromosome aberrations can produce a similar phenotype; therefore these should be excluded by karyotype and microarray before assigning a definitive diagnosis. While sonographic findings are highly variable, common findings include diaphragmatic hernia, cardiovascular malformations, cystic hygroma, and polyhydramnios. Clinical presentation is largely dependent on the particular anomalies present and organ systems affected. The mortality rate for in utero demise combined with early neonatal death is extremely high; however, a small number of patients have survived into childhood with varying severity of clinical manifestations. Careful consideration to the diagnosis of this syndrome should be paid given its poor prognosis and increased recurrence risk to subsequent pregnancies.
Keywords
Fryns syndrome, congenital diaphragmatic hernia, distal limb hypoplasia, cystic hygroma, pulmonary hypoplasia
Introduction
First described in 1979, Fryns syndrome is a rare autosomal recessive disorder characterized by diaphragmatic defects, dysmorphic facial features, distal limb hypoplasia, pulmonary hypoplasia, and associated anomalies of other major organ systems. To date the diagnosis is made solely by clinical criteria, as only one candidate gene has been identified through whole exome sequencing. Although Fryns syndrome was initially thought to be uniformly lethal, there are now multiple reports of survivors exhibiting varying phenotypes. While prenatal ultrasound (US) may identify features suggestive of Fryns syndrome, postnatal confirmation is required for diagnosis.
Disorder
Definition
Diagnostic guidelines have most recently been reviewed and revised by Lin et al. in 2005 and include six major clinical features :
- 1.
diaphragmatic defect (hernia, eventration, hypoplasia, agenesis)
- 2.
distal digital hypoplasia (nails and/or phalanges)
- 3.
significant pulmonary hypoplasia
- 4.
characteristic facial appearance (coarse face, hypertelorism, broad and flat nasal bridge, long philtrum, low-set and poorly formed ears, tented upper lip with macrostomia, micrognathia)
- 5.
associated anomalies (polyhydramnios, cloudy cornea or microphthalmia, orofacial cleft, brain malformation, cardiovascular malformation, renal dysplasia, gastrointestinal malformation, genitourinary malformation)
- 6.
sibling affected with Fryns syndrome
- 1.
Narrowly defined: four of six clinical features.
- 2.
Broadly defined: three of six clinical features.
- 3.
Atypical.
Prevalence and Epidemiology
Over 110 cases of Fryns syndrome have been reported since its initial description in 1979, with an estimated prevalence of 7 : 100,000 live births. It is the most common syndrome associated with congenital diaphragmatic hernia, with a risk association ranging from 1%–10%.
Etiology and Pathophysiology
Fryns syndrome is inherited in an autosomal recessive pattern and should be considered in any patient with a congenital diaphragmatic hernia and a prior affected family member. Although the molecular mechanism has yet to be elucidated, multiple chromosome aberrations, such as microdeletions on chromosomes 1, 8, and 15; partial trisomy 22; and mosaic tetrasomy 12p, have been associated with a similar phenotype. It is recommended that these chromosome abnormalities be excluded by karyotype or chromosome microarray before assigning the diagnosis of Fryns syndrome.
The pathogenesis of Fryns syndrome is not well understood; however, abnormal neural crest cell migration may play a role. In addition, with the clinical implementation of whole exome sequencing, novel deleterious mutations in the PIGN gene have been reported in multiple individuals with a clinical diagnosis of Fryns syndrome. PIGN is one of a number of genes that codes for the production of glycosylphosphatidylinositol (GPI), which functions by attaching various proteins to the lipid layer of cell membranes. GPIs are involved in many cellular processes, some of which function in developmental pathway signaling. Mutations in this gene have also been reported in other individuals with multiple congenital anomalies. Additional research is needed to delineate the association between PIGN and Fryns syndrome as well as to determine other genes that may be implicated in this condition.
Manifestations of Disease
Clinical Presentation
The clinical presentation of Fryns syndrome is largely dependent on the particular anomalies present and the organ systems affected by these anomalies. As congenital diaphragmatic hernia is a common finding, respiratory distress and/or failure secondary to pulmonary hypoplasia, is often present in early neonatal life. The mortality rate for in utero death combined with early neonatal death has been quoted to be as high as 86%. Surviving patients have exhibited a range of clinical manifestations including seizures, developmental delay, and mild to severe intellectual disability. Both postnatal exam as well as pathologic exam at the time of autopsy may reveal clinical features such as facial dysmorphism, hypoplastic thorax with widely spaced nipples, cloudy cornea, nail hypoplasia, brachytelephalangy, webbed neck, gastroesophageal reflux, Hirschsprung disease, and hydronephrosis with vesicoureteral reflux. The most common cardiac malformations observed in Fryns syndrome include conotruncal and aortic arch defects, and both atrial and ventricular septal defects.
Imaging Technique and Findings
Ultrasound.
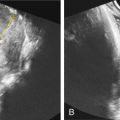
Although not required for diagnosis, diaphragmatic hernia in the presence of another major anomaly should prompt concern for Fryns syndrome ( Figs. 129.1 and 129.2 ). Diaphragmatic hernias observed in Fryns syndrome tend to be posterolateral defects associated with significant lung hypoplasia. Other prenatal sonographic findings may include the following:
- •
ventriculomegaly
- •
Dandy-Walker malformation
- •
agenesis of the corpus callosum
- •
hypoplasia of the distal phalanges
- •
renal malformations (bilateral renal enlargement and/or cystic kidneys)
- •
intestinal malrotation
- •
micrognathia
- •
cleft lip/palate
- •
cardiovascular malformations (most commonly conotruncal defects, aortic arch defects, and atrial/ventricular septal defects).