Cerebrospinal vascular malformations are a group of anomalies affecting the arterial wall, the capillary arteriovenous interface, or the venous and lymphatic structures. Heritability and family studies allow identification of mutations in single genes associated with rare familial conditions causing cerebral or spinal vascular malformations, as is the case in hemorrhagic hereditary telangiectasia diseases. This article reviews the genetic and epigenetic influences increasingly reported in recent years as causal factors or triggers involved in the formation and growth of cerebromedullary vascular malformations.
Key points
- •
Cerebral vascular malformations include anomalies of the arterial wall (ie, cerebral aneurysm), anomalies of the capillary–venous interface (ie, arteriovenous malformation), and venous anomalies (ie, cavernous malformations).
- •
Familial disorders caused by mutations in single genes are associated with cerebral aneurysms (polycystic renal disease), arteriovenous malformations (hereditary disorders such as hemorrhagic hereditary telangiectasia), and cavernous malformations.
- •
Recent studies identified critical genes involved in the development of sporadic forms of the same vascular disorders, emphasizing the role played by genetic and epigenetic factors in their genesis and evolution.
- •
Identification of genetic markers and therapeutic tools and strategies directed to alter the molecular pathways of vascular disorders are in the scope of future research.
Aneurysms
The term aneurysm encompasses a variety of lesions affecting the arterial vascular wall through different mechanisms, leading some investigators to use the term, aneurysmal vasculopathy , which allows categorization of these lesions in different subgroups. Aneurysms result from luminal, parietal, or abluminal injuries or from a combination of all of these.
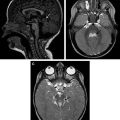
Endothelial or adventitial chronic focal dissections causing recurrent mural hematomas, intramural thrombosis, intrathrombotic channels, and inflammatory changes in the adventitia are seen in serpentine or partially thrombosed aneurysms ( Fig. 1 ). Giant aneurysms (larger than 2.5 cm) without intralesional thrombosis should be considered a type of focal dissections associated with failed angiogenesis in their walls ( Fig. 2 ). Proteins involved in the biological functions of proliferation, development, and apoptosis are highly expressed in large and giant unruptured aneurysms but scarcely expressed in unruptured aneurysms, suggesting that certain proteins are associated with aneurysm growth but not with their ruptures. Giant, serpentine, fusiform, and partially thrombosed aneurysms are associated with mural inflammatory processes. High-resolution black-blood contrast-enhanced T1-weighted magnetic resonance (MR) sequences can be used to detect macrophage activity and inflammation in the arterial walls and show focal contrast enhancement of the walls of inflamed aneurysms ( Fig. 3 ). Patients with familial or acquired disorders mediated by immunologic pathways, such as familial candidiasis, or patients with the acquired immunodeficiency syndrome, may harbor multiple fusiform dysplastic aneurysms, but the responsible molecular mechanisms are unknown. Other inherited disorders, such as neurofibromatosis type 1 ( OMIM# 162200 autosomal dominant), and connective tissue diseases such as Ehlers-Danlos syndrome type IV ( OMIM #130050 , autosomal dominant) and Marfan syndrome ( OMIM #154700 , autosomal dominant) are associated with dysplastic vascular segments and fusiform or giant aneurysms. Noninflammatory conditions such as fibromuscular dysplasia are also associated with intracranial aneurysms (IA), and although their cause is unknown, there have been reports of familial cases. Fusiform and large aneurysms may also be associated with cardiac myxoma in both the sporadic and familiar autosomal dominant forms.
Most of the research on genetics of aneurysms is focused on the study of formation, growth, and rupture of saccular aneurysms or “berry aneurysms” ( Fig 4 ). Saccular intracranial aneurysms are defined as vascular out pouches located mainly around the circle of Willis, usually measuring less than 25 mm (with most ruptured ones measuring 5–7 mm), more frequent in women between 40 and 60 years, very infrequent in pediatric populations, and associated to risk factors such as high blood pressure, cigarette smoking, and alcohol consumption. They are considered the most frequent type of aneurysm, accounting for 80% to 90% of all IA; however, the term saccular aneurysm is not used homogenously in the literature. In a recent meta-analysis, the prevalence of saccular IA was estimated at 3.2% of patients without comorbidities, whereas the prevalence was 6.9% in patients with Autosomal dominant polycystic kidney disease (ADPKD) and 3%–4% in patients with a positive family history of IA or subarachnoid hemorrhage. The same investigators point out that the high incidence of subarachnoid hemorrhage in Finland and Japan does not correlate with a higher incidence of IA but rather with a high rate of rupture. ADPKD is a genetic disorder caused by mutations in 2 genes, PDK1 and PDK2, accounting for 85% to 90% and 10% to 15% of cases, respectively, and characterized by multiple renal cysts leading to progressive renal failure and hypertension. Cerebral aneurysms in ADPKD are 5 times more frequent than those found in the general population, and the average age for their rupture is 41 years, a decade earlier than in the general population. IA in ADPKD is probably not related to early onset of hypertension. Recessive or infantile forms of PKD are not associated with IA. Proteins involved in ADPKD are expressed in vascular smooth muscle and endothelium, suggesting that they have a direct role in the vascular manifestations of ADPKD. ADPKD accounts for 9% of familial IA. The tuberous sclerosis complex ( OMIM#191100 ) is an autosomal dominant disorder caused by mutations in 2 genes (TSC1 and TSC2). TSC2 is located immediately adjacent to PDK1, and large deletions could result in a contiguous gene syndrome. IA associated with tuberous sclerosis complex is thought to be a part of a continuous gene syndrome ( Fig. 5 ).
In the absence of any heritable disorder, the prevalence of IA in first-degree relatives of patients with IA is estimated at 6% to 20%. The Familial Aneurysm study, a multicentric study comparing the characteristics of familiar and nonfamilial IA in more than 3000 participants found that familial IA are more likely multiple, are more likely to be located in the middle cerebral artery, and present higher risk of rupture than sporadic IA. Similar data are reported in several other population-based and non–population-based studies.
It seems that genetic factors likely influence the occurrence of IA in association with hemodynamic factors or modifiable risk factors such as hypertension, alcohol abuse, and cigarette smoking. Candidate gene studies in sporadic IA have identified several potential risk loci with the affected proteins involved in the regulation of the extracerebral matrix, endothelium maintenance, and the inflammatory pathways through the regulation of macrophages, lymphocytes, and interleukins. More recently, genomewide association studies (GWAS) using large cohorts of affected and nonaffected individuals are trying to identify variations in the entire genome (instead of in a candidate gene) that are common in individuals with a particular trait but not in the general population. Using this technology, several loci associated to IA were identified, and the loci 9p21.3, which is involved in multiple molecular pathways of cellular proliferation, seems to be the most consistent and significant one associated with IA. Genes on chromosomes 8, 4, 10, 13, and 18 expressed in smooth muscle and endothelial cells and collectively involved in regulating the cell cycle progression, cellular proliferation, and mechanisms of vasculogenesis and angiogenesis are also thought to be involved in the mechanism of IA formation and growth. A recent meta-analysis of all genetic studies in IA including candidate gene studies and GWAS in 116,000 individuals found that multiple pathologic pathways involved in vascular endothelial maintenance and extracellular matrix integrity are present in the formation and rupture of IA, confirming that variants on chromosomes 4, 8, and 9 are strongly associated with IA. Interesting work is ongoing and focused on identifying molecular markers in peripheral blood to predict the risk of rupture, occurrence, and development of IA.
The annual risk of rupture of IA is estimated at about 1%. More than 30,000 subarachnoid hemorrhages (SAH) caused by ruptured IA are diagnosed every year in the United States. Mortality associated with IA rupture is estimated at 40% to 50% and with two-thirds of survivors having permanent neurologic deficits. However, most of IA will not rupture. Identification of risk factors and mechanisms leading to aneurysms rupture is one of the goals of current research. The next generation of genetics tools and mega-analysis studies with high power of predictions will likely help us understand the mechanisms of formation and rupture of IA and the links between genetic and environmental factors.
Guidelines from the American Heart Association published in 2012 consider it reasonable to offer noninvasive screening to patients with familial (at least 1 first-degree relative) SAH and/or a history of SAH to evaluate for de novo aneurysms or late regrowth of treated aneurysms, but the risks and benefits of this screening still require further study (class IIb; level of evidence B). Based on a cost-effectiveness study, Bor and colleagues consider it acceptable to screen patients with 2 or more affected first-degree relatives. Screening should start at 20 years of age and be repeated every 7 years up to 80 years.
Digital subtraction angiography (DSA) is the gold standard to detect IA, but it carries a 0.5% risk of permanent neurologic sequelae. Noninvasive imaging techniques (computed tomography angiography/magnetic resonance [MR] imaging) should be considered the first option for screening of high-risk asymptomatic populations.
Aneurysms
The term aneurysm encompasses a variety of lesions affecting the arterial vascular wall through different mechanisms, leading some investigators to use the term, aneurysmal vasculopathy , which allows categorization of these lesions in different subgroups. Aneurysms result from luminal, parietal, or abluminal injuries or from a combination of all of these.
Endothelial or adventitial chronic focal dissections causing recurrent mural hematomas, intramural thrombosis, intrathrombotic channels, and inflammatory changes in the adventitia are seen in serpentine or partially thrombosed aneurysms ( Fig. 1 ). Giant aneurysms (larger than 2.5 cm) without intralesional thrombosis should be considered a type of focal dissections associated with failed angiogenesis in their walls ( Fig. 2 ). Proteins involved in the biological functions of proliferation, development, and apoptosis are highly expressed in large and giant unruptured aneurysms but scarcely expressed in unruptured aneurysms, suggesting that certain proteins are associated with aneurysm growth but not with their ruptures. Giant, serpentine, fusiform, and partially thrombosed aneurysms are associated with mural inflammatory processes. High-resolution black-blood contrast-enhanced T1-weighted magnetic resonance (MR) sequences can be used to detect macrophage activity and inflammation in the arterial walls and show focal contrast enhancement of the walls of inflamed aneurysms ( Fig. 3 ). Patients with familial or acquired disorders mediated by immunologic pathways, such as familial candidiasis, or patients with the acquired immunodeficiency syndrome, may harbor multiple fusiform dysplastic aneurysms, but the responsible molecular mechanisms are unknown. Other inherited disorders, such as neurofibromatosis type 1 ( OMIM# 162200 autosomal dominant), and connective tissue diseases such as Ehlers-Danlos syndrome type IV ( OMIM #130050 , autosomal dominant) and Marfan syndrome ( OMIM #154700 , autosomal dominant) are associated with dysplastic vascular segments and fusiform or giant aneurysms. Noninflammatory conditions such as fibromuscular dysplasia are also associated with intracranial aneurysms (IA), and although their cause is unknown, there have been reports of familial cases. Fusiform and large aneurysms may also be associated with cardiac myxoma in both the sporadic and familiar autosomal dominant forms.
Most of the research on genetics of aneurysms is focused on the study of formation, growth, and rupture of saccular aneurysms or “berry aneurysms” ( Fig 4 ). Saccular intracranial aneurysms are defined as vascular out pouches located mainly around the circle of Willis, usually measuring less than 25 mm (with most ruptured ones measuring 5–7 mm), more frequent in women between 40 and 60 years, very infrequent in pediatric populations, and associated to risk factors such as high blood pressure, cigarette smoking, and alcohol consumption. They are considered the most frequent type of aneurysm, accounting for 80% to 90% of all IA; however, the term saccular aneurysm is not used homogenously in the literature. In a recent meta-analysis, the prevalence of saccular IA was estimated at 3.2% of patients without comorbidities, whereas the prevalence was 6.9% in patients with Autosomal dominant polycystic kidney disease (ADPKD) and 3%–4% in patients with a positive family history of IA or subarachnoid hemorrhage. The same investigators point out that the high incidence of subarachnoid hemorrhage in Finland and Japan does not correlate with a higher incidence of IA but rather with a high rate of rupture. ADPKD is a genetic disorder caused by mutations in 2 genes, PDK1 and PDK2, accounting for 85% to 90% and 10% to 15% of cases, respectively, and characterized by multiple renal cysts leading to progressive renal failure and hypertension. Cerebral aneurysms in ADPKD are 5 times more frequent than those found in the general population, and the average age for their rupture is 41 years, a decade earlier than in the general population. IA in ADPKD is probably not related to early onset of hypertension. Recessive or infantile forms of PKD are not associated with IA. Proteins involved in ADPKD are expressed in vascular smooth muscle and endothelium, suggesting that they have a direct role in the vascular manifestations of ADPKD. ADPKD accounts for 9% of familial IA. The tuberous sclerosis complex ( OMIM#191100 ) is an autosomal dominant disorder caused by mutations in 2 genes (TSC1 and TSC2). TSC2 is located immediately adjacent to PDK1, and large deletions could result in a contiguous gene syndrome. IA associated with tuberous sclerosis complex is thought to be a part of a continuous gene syndrome ( Fig. 5 ).
In the absence of any heritable disorder, the prevalence of IA in first-degree relatives of patients with IA is estimated at 6% to 20%. The Familial Aneurysm study, a multicentric study comparing the characteristics of familiar and nonfamilial IA in more than 3000 participants found that familial IA are more likely multiple, are more likely to be located in the middle cerebral artery, and present higher risk of rupture than sporadic IA. Similar data are reported in several other population-based and non–population-based studies.
It seems that genetic factors likely influence the occurrence of IA in association with hemodynamic factors or modifiable risk factors such as hypertension, alcohol abuse, and cigarette smoking. Candidate gene studies in sporadic IA have identified several potential risk loci with the affected proteins involved in the regulation of the extracerebral matrix, endothelium maintenance, and the inflammatory pathways through the regulation of macrophages, lymphocytes, and interleukins. More recently, genomewide association studies (GWAS) using large cohorts of affected and nonaffected individuals are trying to identify variations in the entire genome (instead of in a candidate gene) that are common in individuals with a particular trait but not in the general population. Using this technology, several loci associated to IA were identified, and the loci 9p21.3, which is involved in multiple molecular pathways of cellular proliferation, seems to be the most consistent and significant one associated with IA. Genes on chromosomes 8, 4, 10, 13, and 18 expressed in smooth muscle and endothelial cells and collectively involved in regulating the cell cycle progression, cellular proliferation, and mechanisms of vasculogenesis and angiogenesis are also thought to be involved in the mechanism of IA formation and growth. A recent meta-analysis of all genetic studies in IA including candidate gene studies and GWAS in 116,000 individuals found that multiple pathologic pathways involved in vascular endothelial maintenance and extracellular matrix integrity are present in the formation and rupture of IA, confirming that variants on chromosomes 4, 8, and 9 are strongly associated with IA. Interesting work is ongoing and focused on identifying molecular markers in peripheral blood to predict the risk of rupture, occurrence, and development of IA.
The annual risk of rupture of IA is estimated at about 1%. More than 30,000 subarachnoid hemorrhages (SAH) caused by ruptured IA are diagnosed every year in the United States. Mortality associated with IA rupture is estimated at 40% to 50% and with two-thirds of survivors having permanent neurologic deficits. However, most of IA will not rupture. Identification of risk factors and mechanisms leading to aneurysms rupture is one of the goals of current research. The next generation of genetics tools and mega-analysis studies with high power of predictions will likely help us understand the mechanisms of formation and rupture of IA and the links between genetic and environmental factors.
Guidelines from the American Heart Association published in 2012 consider it reasonable to offer noninvasive screening to patients with familial (at least 1 first-degree relative) SAH and/or a history of SAH to evaluate for de novo aneurysms or late regrowth of treated aneurysms, but the risks and benefits of this screening still require further study (class IIb; level of evidence B). Based on a cost-effectiveness study, Bor and colleagues consider it acceptable to screen patients with 2 or more affected first-degree relatives. Screening should start at 20 years of age and be repeated every 7 years up to 80 years.
Digital subtraction angiography (DSA) is the gold standard to detect IA, but it carries a 0.5% risk of permanent neurologic sequelae. Noninvasive imaging techniques (computed tomography angiography/magnetic resonance [MR] imaging) should be considered the first option for screening of high-risk asymptomatic populations.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


