Outline
Chromosomal Defects, 27
Abnormalities of Chromosome Number, 27
Trisomy 21, 27
Trisomy 18, 28
Trisomy 13, 28
Turner Syndrome, 28
Klinefelter Syndrome, 28
Triploidy, 29
Abnormalities of Chromosome Structure, 29
Other Types of Chromosomal Rearrangements, 29
Single Gene Disorders, 29
Autosomal-Dominant Inheritance, 31
Penetrance and Expression, 31
Autosomal-Recessive Disorders, 32
X-Linked Inheritance, 32
Other Novel Genetic Mechanisms, 32
Multifactorial Inheritance, 33
Teratology, 34
Recognized Teratogenic Agents, 35
Maternal Infections, 35
Medications, 35
Radiation, 35
Maternal Factors, 35
Mechanical Factors, 37
Genetic Counseling, 37
Genetic Screening in Pregnancy, 37
Carrier (Heterozygote) Screening for Single Gene Disorders, 38
Screening for Structural Birth Defects, 40
Alpha-Fetoprotein and Open Neural Tube Defects, 40
Elevated MSAFP, 41
Other Abnormalities and Elevated AFP, 41
Fetal Chromosomal Aneuploidy, 41
First Trimester Risk Assessment, 41
Second Trimester Risk Assessment, 42
Sonographic “Soft Markers” for Aneuploidy, 43
Combined First and Second Trimester Risk Assessment, 43
Cell Free DNA Screening, 43
Prenatal Screening in Multiple Gestations, 45
Third-Trimester Pregnancy Complications After Abnormal Serum Screening, 45
Genetic Diagnosis in Pregnancy, 45
Genetic Counseling Before Prenatal Diagnostic Procedures, 46
Techniques for Prenatal Diagnosis, 46
Multiple Gestations, 47
Postprocedural Loss Rates in Multiple Gestations, 48
Selective Termination of Multifetal Pregnancies, 48
Fetal Blood Sampling and Other Fetal Tissue Biopsy, 48
Preimplantation Genetic Diagnosis, 49
Prenatal Testing for Congenital Malformations, 49
Techniques of Genetic Analysis, 50
Chromosome Identification, 50
Fluorescence in Situ Hybridization, 51
Molecular Techniques for DNA Analysis, 51
Chromosomal Microarray, 51
Summary of Key Points
- •
In the population, 2% to 3% of newborns have a congenital malformation or genetic disease identified at birth.
- •
Despite advances in genetics, the cause of more than half of human congenital abnormalities remains unknown.
- •
Chromosomal abnormalities are present in about 0.9% of newborns and include abnormalities of chromosome number as well as abnormalities of chromosome structure.
- •
The embryo is most sensitive to teratogenic effects between 3 and 8 weeks of development.
- •
Current available tools for fetal aneuploidy screening include cell-free DNA screening, various forms of multiple marker screening with first and second trimester maternal serum analytes, and ultrasound measurements, including nuchal translucency.
- •
The prevalence of many single gene disorders varies with race and ethnicity, and testing is often recommended based on an individual patient’s background.
- •
Expanded carrier screening, including panels to simultaneously test for a large number of genetic conditions, is increasingly utilized for prenatal genetic screening.
- •
Chorionic villus sampling (CVS) and amniocentesis are both routinely used for prenatal diagnostic testing and can provide tissue for such tests as fluorescence in situ hybridization (FISH), karyotyping, chromosomal microarray analysis (CMA), and DNA-based tests.
- •
The loss rate attributable to CVS and amniocentesis is estimated to be between 1/500 and 1/1000 and decreases with provider experience.
- •
Many structural fetal abnormalities are associated with an increased risk of aneuploidy as well as copy number variants detectable with chromosomal microarray.
Genetic diseases are often perceived to be so rare that the average practitioner will seldom encounter them. However, increasing knowledge and technologic advances in genetic testing have demonstrated that this is far from the case. The availability of prenatal diagnosis for a wide range of disorders continues to increase with advances in genetics. In addition, progress has been made in population screening tests to identify couples who carry a genetic disorder. New techniques, such as cell-free DNA screening, have also changed the field of prenatal diagnosis significantly. These improvements in prenatal screening and diagnosis mean that many more at-risk couples are able to have unaffected children. In addition to reproductive choice, carrier screening and fetal diagnostic testing afford the important opportunity for preparation of the family and the delivery site for the birth of a fetus with a known genetic disorder.
Ultrasound plays a central role in the provision of prenatal screening and diagnosis. Not only is ultrasound key to guiding prenatal diagnostic procedures, but also integration of a genetics-based prenatal diagnosis program has been shown to increase the accuracy of diagnosis when compared to ultrasound alone. This chapter includes a discussion of genetics, with an emphasis on recent advances relevant to prenatal diagnosis and a description of current strategies for genetic testing with a focus on how genetic screening and sonography together contribute to the provision of accurate and precise prenatal diagnosis.
Genetics and Birth Defects
According to most studies, 2% to 3% of living newborns have a congenital malformation. When considering birth defects noted in the first years of life, this incidence is nearly doubled. With the decline in infant mortality in the United States from infection and malnutrition, congenital malformations are now a leading cause of infant death (>20%) and are responsible for greater than 30% of intensive care nursery admissions. Congenital defects range from enzyme deficiencies caused by single gene mutations to complex associations of structural defects. The continuum between purely biochemical abnormalities and structural birth defects includes disorders of structure, function, metabolism, and behavior.
Birth defects result from the interaction between the genetic makeup of the embryo and the environment in which it develops. The basic developmental information is encoded in genes, but the genotype is subjected to environmental influences that can impact the observed phenotype. In some cases, the genetic information is expressed regardless of environment, whereas in others, environmental causes interfere with normal development despite a normal genotype. Although some processes are primarily environmental and others mainly genetic, the distinctions between the two are not perfect.
Despite considerable advances and research over past several decades, the cause of more than half of human congenital abnormalities remains unknown. Of those with a recognized cause, approximately 15% to 20% are autosomal genetic diseases and 20% are cytogenetic in origin. Fewer than 1% of anomalies are thought to result from teratogenic medications. Some of the remaining defects are associated with other environmental exposures during pregnancy, including infectious agents (3%), maternal disease states (4%), mechanical problems (1% to 2%), irradiation, and unknown environmental causes. The remainder are of unknown or complex causes (multifactorial, polygenic, spontaneous errors of development, and synergistic interactions of teratogens) ( Fig. 2-1 ).
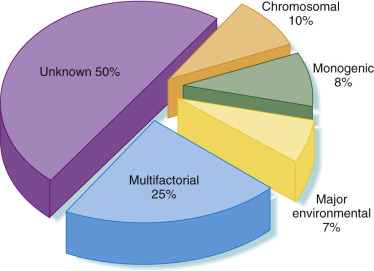
Developmental Disorders: Causes, Mechanisms, and Patterns
Errors in morphogenesis are often classified by dysmorphologists according to the underlying pathogenesis ( Fig. 2-2 ). Malformations are defects in the structure of an organ resulting from a specific primary abnormality of development, such as a congenital heart or neural tube defect (NTD). Deformations are abnormalities of form, shape, or position caused by mechanical forces such as intrauterine molding or constraint. Factors leading to deformations may be extrinsic (e.g., oligohydramnios owing to ruptured membranes) or intrinsic (e.g., oligohydramnios owing to renal agenesis). Deformations may also occur postnatally; for example, an infant may develop a flat head from sleeping in one position. A disruption is a morphologic defect that results from breakdown of previously normal tissue. Disruptions can be due to extrinsic forces, internal interferences with a developmental process, or vascular insults. Examples of disruptions include amputations owing to amniotic bands, and gastroschisis and porencephaly, both thought to result from in utero vascular insults.
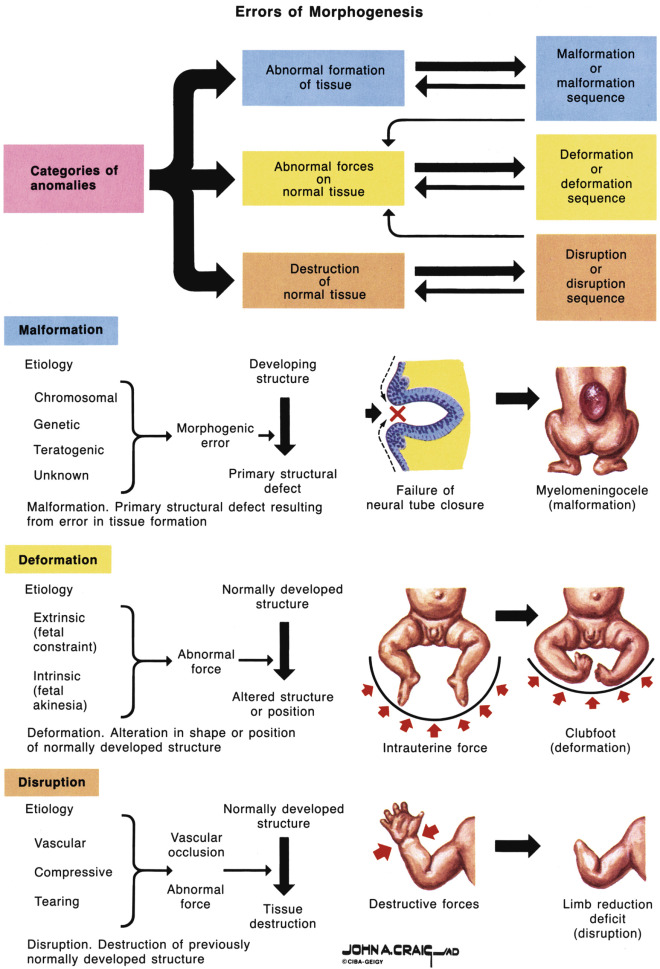
A sequence is a pattern of multiple abnormalities resulting from a single primary anomaly or mechanical factor; it may be a malformation, deformation, or disruption. An example is Potter sequence, in which oligohydramnios from any cause leads to similar features of fetal compression: characteristic facial features and abnormal positioning of the hands and feet. A syndrome is a pattern of multiple abnormalities known to have a common, specific cause. An example is Down syndrome (DS), which is caused by the presence of an extra copy of chromosome 21. Finally, the term association is used when two or more features occur together more commonly than expected by chance alone but for which no cause has been demonstrated. A common example is VACTERL association (Vertebral defects, Anal atresia, Cardiac defects, Tracheoesophageal fistula with Esophageal atresia, Renal anomalies, and Limb anomalies). This term does not imply a specific underlying cause but rather prompts the search for specific other defects when one component of the association is identified.
Chromosomal Defects
Chromosomal abnormalities occur in approximately 0.9% of newborns, and it is estimated that at least 10% to 15% of conceptions are chromosomally abnormal. Chromosome abnormalities are the leading cause of pregnancy loss, and at least 95% of chromosomally abnormal conceptions are lost before term. Abnormalities seen in abortuses differ from those seen in liveborn infants because the most severe chromosome abnormalities result in early arrest of development, whereas less severe abnormalities yield milder phenotypes that allow survival until later in pregnancy or even live birth. The most common abnormalities seen in first trimester spontaneous abortions are 45,X and 47,+16 (trisomy 16). Full or complete trisomy 16 is never seen in live births, whereas fewer than 1% of conceptions with 45,X survive until term.
Chromosome abnormalities can be either numeric or structural. They can involve one or more autosomes (chromosomes numbered 1-22), sex chromosomes, or both simultaneously. Aneuploidy refers to the presence of an abnormal number of chromosomes and is almost always associated with abnormalities of physical and cognitive development. Translocations , the exchange of segments between nonhomologous chromosomes, are relatively common and can be balanced, in which the appropriate amount of chromosomal material is present but rearranged, or unbalanced, in which some chromosomal material is gained or lost. Individuals with a balanced translocation are most often phenotypically normal unless the break for the rearrangement occurs in a critical gene and disrupts its function. Those with an unbalanced rearrangement most often have some phenotypic consequence including intellectual disability and structural birth defects as a result of being trisomic for one segment and monosomic for another. Many autosome translocations are unique, with some variation in expression of the additional or missing genetic material. A Robertsonian translocation is a rearrangement that involves any of the five acrocentric chromosomes, namely, 13, 14, 15, 21, and 22. The incidence of balanced Robertsonian translocations in the general population is 1/1000 and confers a higher risk for the relevant trisomies (most commonly trisomy 21 and 13) in offspring of balanced carriers. Balanced carriers are phenotypically normal and often only become aware of the rearrangement when they experience pregnancy with an abnormal karyotype, recurrent miscarriages, or male infertility.
Abnormalities of Chromosome Number
Aneuploidy is the most common clinically significant type of human chromosomal abnormality and occurs in 3% to 4% of recognized pregnancies. Nondisjunction in mitosis or meiosis is the cause of most aneuploidies. Maternal meiotic nondisjunction is known to increase with maternal age, and therefore the risk for aneuploid offspring also increases with advancing maternal age ( Table 2-1 ). Both trisomy, the presence of three copies of an individual chromosome, and monosomy, the presence of a single copy of a chromosome, typically have significant phenotypic consequences. Monosomy is seen less frequently than trisomy because this situation is generally not compatible with life. Trisomy or monosomy for any chromosome can occur theoretically, but in practice, some are seen much more frequently than others ( Table 2-2 ). This is because most aneuploidies are incompatible with life, and aneuploid embryos spontaneously abort very early in gestation. Some chromosome abnormalities, for example, trisomy 8, are seen at birth only in mosaic form, and the full trisomy is likely lethal. Mosaicism is defined as the presence of two different cell lines with different genotypes in the same individual. Mosaicism is not always deleterious but can result in an abnormal phenotype depending on the degree and type of affected tissue. An entire extra set or sets of chromosomes, polyploidy, is also possible but is not compatible with long-term survival.
| Maternal Age (Years) | Risk for Down Syndrome | Total Risk for Chromosomal Abnormalities * |
|---|---|---|
| 20 | 1/1667 | 1/526 |
| 21 | 1/1667 | 1/526 |
| 22 | 1/1429 | 1/500 |
| 23 | 1/1429 | 1/500 |
| 24 | 1/1250 | 1/476 |
| 25 | 1/1250 | 1/476 |
| 26 | 1/1176 | 1/476 |
| 27 | 1/1111 | 1/455 |
| 28 | 1/1053 | 1/435 |
| 29 | 1/1000 | 1/417 |
| 30 | 1/952 | 1/384 |
| 31 | 1/909 | 1/384 |
| 32 | 1/769 | 1/323 |
| 33 | 1/625 | 1/286 |
| 34 | 1/500 | 1/238 |
| 35 | 1/385 | 1/192 |
| 36 | 1/294 | 1/156 |
| 37 | 1/227 | 1/127 |
| 38 | 1/175 | 1/102 |
| 39 | 1/137 | 1/83 |
| 40 | 1/106 | 1/66 |
| 41 | 1/82 | 1/53 |
| 42 | 1/64 | 1/42 |
| 43 | 1/50 | 1/33 |
| 44 | 1/38 | 1/26 |
| 45 | 1/30 | 1/21 |
| 46 | 1/23 | 1/16 |
| 47 | 1/18 | 1/13 |
| 48 | 1/14 | 1/10 |
| 49 | 1/11 | 1/8 |
| Disorder | Frequency per 1000 Live Births * |
|---|---|
| Trisomy 21 | 1.5 |
| Trisomy 18 | 0.12 |
| Trisomy 13 | 0.07 |
| 47,XXY (Klinefelter syndrome) | 1.5 |
| 45.X (Turner syndrome) | 0.4 |
| XYY syndrome | 1.5 |
| XXX syndrome | 0.65 |
* Births of appropriate sex only for sex chromosomal abnormalities.
Trisomy 21
The most common aneuploidy detected at birth is trisomy 21, which occurs in approximately 1/700 live births ( Fig. 2-3 ). Trisomy 21 occurs as a result of maternal nondisjunction during meiosis in 95% of cases of DS; the remainder of cases is due to translocations involving chromosome 21. Karyotype analysis from the affected individual can differentiate between nondisjunction and translocation causes. Individuals with DS from either cause have a characteristic phenotype that includes distinctive facial features including midface hypoplasia and upslanting palpebral fissures, short stature, brachycephaly, a short neck with redundant skin on the nape, short broad hands with a single transverse palmar crease, and hypotonia. Other findings include intellectual disability, an increased risk for acute myeloid leukemia in the first 3 years of life, and thyroid dysfunction. Congenital heart defects (CHDs) are reported in 44% to 58% of affected newborns and are important contributors to morbidity and fatality. Other structural birth defects associated with this diagnosis include hydronephrosis and duodenal atresia.

The recurrence risk of trisomy 21 (or other autosomal trisomies) is approximately 1% after one such child with nondisjunction-related aneuploidy is born to a couple. Although often requested, there is no indication for parental chromosomal evaluation after the birth of a child with trisomy 21 unless it is due to a translocation. A history of DS elsewhere in the family does not increase the risk of having a child with a chromosome abnormality, provided there is no familial translocation.
Trisomy 18
Trisomy 18 occurs in about 1/8000 live births but is much more common at conception, with about 95% of such cases resulting in spontaneous abortion or stillbirth. Postnatal survival is poor, and the majority of liveborn neonates dies in early infancy. Characteristic findings include cardiac malformations, severe intellectual disability, growth restriction, a prominent occiput, dolicocephaly, small mandible, short sternum, clenched hands with overlapping digits, and rocker-bottom feet. Choroid plexus cysts are identified more frequently in this population, although most fetuses that demonstrate choroid plexus cysts are not aneuploid.
Trisomy 13
Trisomy 13 occurs in about 1/20,000 live births. There is a high stillbirth rate among pregnancies with this diagnosis, and those who are liveborn typically die in early infancy. The phenotype includes midline defects including severe central nervous system (CNS) malformations such as holoprosencephaly, severe intellectual disability, growth restriction, cleft lip and palate, microphthalmia, omphalocele, polydactyly, clenched hands with overlapping digits, CHDs, and renal abnormalities such as polycystic kidneys.
Turner Syndrome (45,X)
Although monosomy for an entire chromosome is almost always lethal, an important exception is monosomy X (45,X), which results in Turner syndrome. Turner syndrome is estimated to occur in 1% to 2% of all conceptions, but most end in miscarriage, resulting in a frequency of 1/2000 to 1/3000 live births. Females with Turner syndrome have a characteristic phenotype, which includes short stature, webbed neck, peripheral lymphedema at birth, CHDs, and renal abnormalities. Other features include ovarian dysgenesis with failure to develop secondary sexual characteristics and associated infertility. Prenatally, fetuses with Turner syndrome often have an increased nuchal translucency or cystic hygroma, lymphangiectasia, structural renal abnormalities, and CHDs, particularly left-sided obstructive lesions such as coarctation of the aorta. Females with Turner syndrome usually have normal intelligence, although they may have specific learning disabilities.
The chromosomal abnormalities that cause Turner syndrome vary. Only about 50% of patients have a nonmosaic 45,X karyotype. Approximately 30% to 40% of cases involve mosaicism, most commonly 45,X/46,XX or 45,X/46,XY. Patients with a Y chromosome may have ambiguous genitalia and are at risk for development of gonadoblastoma in their dysgenetic gonads. Structural abnormalities of the X chromosome are seen in 10% to 20% of cases and often involve mosaicism. The fact that these patients have more than one cell line can account for the variability in phenotype of individuals affected with Turner syndrome. Unlike the other diagnoses discussed, the nondisjunction that most often results in Turner syndrome is paternal in origin and is not associated with the age of either parent. There is no increased risk in subsequent pregnancies for another fetus with a chromosome abnormality.
Klinefelter Syndrome (47,XXY)
Klinefelter syndrome (47,XXY) occurs in about 1/1000 male births and is a common cause of primary hypogonadism. Men with Klinefelter syndrome tend to be tall, with long arms and legs. They have small testes and are sterile owing to atrophy of the seminiferous tubules. Gynecomastia occurs in about 30%. There is an increased risk for learning disabilities, with intelligence quotient (IQ) about 10 to 15 points below that of unaffected siblings, although men with Klinefelter syndrome do not usually have significant intellectual disability. Because the disorder is often subtle, men are often first diagnosed in infertility clinics. This condition is typically not identified by prenatal ultrasound findings because it is not associated with structural birth defects. In the prenatal setting, Klinefelter syndrome is most commonly incidentally identified by diagnostic testing done during pregnancy for another indication.
Triploidy
Triploidy is a type of polyploidy and is defined as an entire extra set of chromosomes (69 chromosomes per cell). Triploidy is generally a lethal condition and is rarely reported in live births. Of those fetuses with triploidy who are liveborn, the longest reported survivors have lived to less than 1 year of age. Congenital abnormalities seen in triploidy include micrognathia, prominent forehead, relative macrocephaly, low-set malformed ears, microphthalmia, hypertelorism, cleft lip and palate, omphalocele, syndactyly, and asymmetric development.
The extra chromosome set in triploid pregnancies can be of either maternal or paternal origin. Maternally derived triploidy results from fertilization of a diploid ovum by a haploid sperm and can involve either an XXX or XXY karyotype. Paternally derived triploidy can result from fertilization of a haploid ovum by either a diploid sperm or by two haploid sperm and can have either an XXX, XXY, or XYY karyotype.
The phenotype of triploid pregnancies depends on the parental source of the extra chromosome set. Triploids with an extra set of paternal chromosomes have a well-formed fetus with an abnormal placenta and may result in partial hydatidiform moles. By definition, all partial molar pregnancies are triploid but not all triploid fetuses have a molar placenta. Those with an extra set of maternal chromosomes usually have a small, growth-restricted fetus and a small, noncystic placenta.
Abnormalities of Chromosome Structure
There are many types of abnormalities of chromosome structure, but all generally result from chromosome breakage, followed by recombination in an abnormal configuration. Overall, structural chromosome abnormalities occur in about 1/375 newborns. Reciprocal translocations involve an exchange of segments between nonhomologous chromosomes. These rearrangements are relatively common and can be balanced (that is, the appropriate amount of chromosomal material is present), typically resulting in a normal outcome, or unbalanced, in which some chromosomal material is gained or lost, therefore resulting in an abnormal outcome. There is an increased risk of unbalanced rearrangements in the offspring of people who carry balanced translocations, thereby leading to decreased fertility and an increased risk of offspring with structural malformations or intellectual disability.
A robertsonian translocation occurs when the long arms of two acrocentric chromosomes (chromosomes 13, 14, 15, 21, and 22) are fused and the corresponding two short arms are lost. Because the short arms of these chromosomes do not contain essential genetic material (they are made up of multiple copies of ribosomal RNA genes), the loss of this material does not result in an abnormal phenotype in a balanced robertsonian translocation carrier. The phenotype of an unbalanced robertsonian translocation is either miscarriage or a child with a trisomic condition such as trisomy 21 or trisomy 13 from the additional material. The most common robertsonian translocation involves chromosomes 14 and 21 and is responsible for about 5% of cases of DS. The recurrence of translocation DS depends on whether the translocation was inherited from a carrier parent. In 75% of cases, the translocation is de novo, and the recurrence risk is very low (<1%). If the translocation was inherited from a parent who carries a 14;21 balanced translocation, there is a 10% chance of recurrence if the mother is the carrier and about a 2% chance if the father is the carrier.
When a balanced translocation or other rearrangement is identified in a fetus at the time of prenatal diagnosis, testing of the parents is recommended. If the translocation was inherited from a phenotypically normal parent, the fetus would be predicted to be normal. If a reciprocal translocation occurs as a de novo event, there is approximately a doubling of the background risk of phenotypic abnormality. This is thought to be due to subtle, undetected chromosomal imbalance or disruption of genes at the breakpoint(s).
Other Types of Chromosomal Rearrangements
In addition to translocations, there are several other types of unbalanced chromosomal rearrangements. These types include deletions , which occur with the loss of a chromosome segment, resulting in partial monosomy. Such deletions can involve one or many genes and are responsible for a number of clinically distinct microdeletion syndromes ( Table 2-3 ). One of the most common is the 22q deletion syndrome, which occurs in about 1/4000 live births. The 22q deletion is a relatively common cause of CHDs, particularly conotruncal abnormalities, such as truncus arteriosus and tetralogy of Fallot. Other features include cleft palate, velopharyngeal incompetence, renal malformations, characteristic facial features, immune deficiency, hypocalcemia, and learning difficulties. The majority of individuals (93%) has a de novo deletion identified by microarray or FISH. When an individual is diagnosed with this condition, parental testing is recommended because of the variable expressivity within a family and the small chance that one of the parents is a more mildly affected carrier.
| Syndrome | Location | Frequency |
|---|---|---|
| Prader-Willi | 15q11-13 | 1/10,000-1/30,000 |
| Angelman | 15q11-13 | 1/12,000-1/20,000 |
| Velocardiofacial, DiGeorge | 22q11.2 | 1/4000 |
| Smith-Magenis | 17p11.2 | 1/15,000-1/25,000 |
| Williams | 7q11.23 | 1/7500 |
| Alagille | 20p12 | 1/30,000-1/50,000 |
| Rubinstein-Taybi | 16p13.3 | 1/100,000 |
| WAGR * | 11p13 | 1/40,000 |
| Miller-Dieker | 17p13.3 | 1/85,000 |
| Wolf-Hirschhorn | 4p16.3 | 1/50,000 |
| Cri-du-chat | 5p15.2 | 1/20,000-1/50,000 |
| Retinoblastoma | 13q14.2 | 1/15,000-1/20,000 |
* Wilms tumor, aniridia, genitourinary anomalies, retardation.
Duplications of a chromosome segment also occur, resulting in partial trisomy. The size and region of the duplication confer risk for abnormal phenotype ranging from normal to significant structural defects and intellectual disability.
Single Gene Disorders
Many important genetic diseases occur as the result of a mutation at a single gene. Gregor Mendel first described the principles of segregation and independent assortment of genes in his well-known experiments with garden peas. This work led to the classic descriptions of autosomal-dominant (AD) and autosomal-recessive (AR) inheritance. X-linked inheritance occurs with conditions encoded on the X chromosome, and it may be either recessive or dominant. Figure 2-4 shows modes of inheritance; Figure 2-5 shows pedigrees demonstrating modes of inheritance.
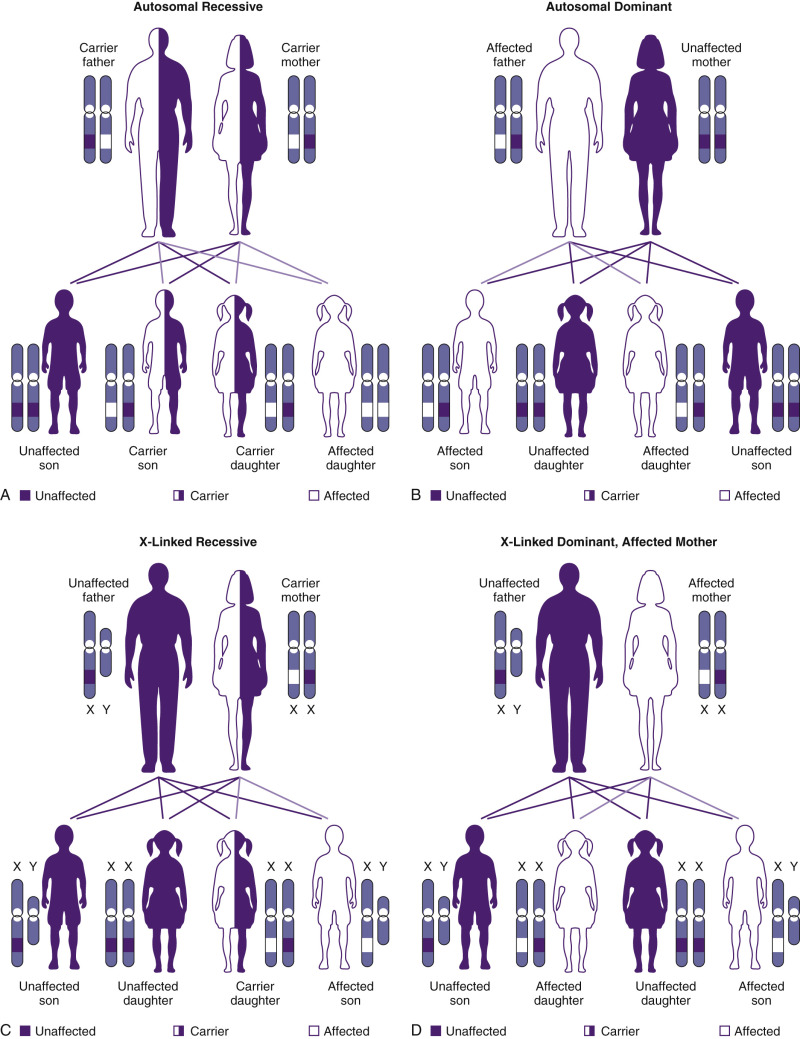
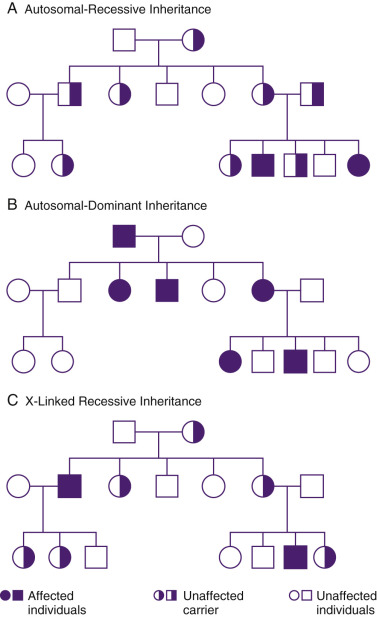
The completion of the human genome project in 2003 was an epic chapter in laying the foundation for the study of human genes and the diagnosis and treatment of genetic disease. Although the human genome has now been sequenced, we still do not know how most genes work in either health or disease. We have gained an increased appreciation, however, for the complexity of human genetics. Relevant to clinical genetics, we have greatly increased our understanding of genetic heterogeneity, and the less than clear-cut association of one gene with one disorder. We now understand that mutations at more than one gene locus can lead to the same phenotype and that multiple different phenotypes can result from mutations in the same gene. Phenotypes also result from the interaction of genotype with the environment (including other genes), and there are numerous phenomena that advances in genetic technology have uncovered that make genetics far more fascinating, if far more complex.
Autosomal-Dominant Inheritance
In AD inheritance, affected individuals are heterozygous for an abnormal allele that they transmit to 50% of their offspring, independent of gender. Overall, approximately 1/200 individuals is affected with an AD condition, although individual conditions are generally rare.
In theory, AD inheritance is straightforward, but in actuality, there are numerous factors affecting gene expression that can make evaluation of such cases extremely complex. Counseling of families regarding AD disorders requires consideration of mechanisms such as the rate of new mutation, mosaicism, conditions with late or variable age of onset, incomplete penetrance, and variation in expressivity. AD disorders may be inherited from an affected parent or may result from a new mutation, with the risk varying with each specific condition. Paternal age has a demonstrable effect on the rate of such new mutations ( Fig. 2-6 ) and is associated with an increased risk for AD disorders, including achondroplasia and neurofibromatosis (NF). No current screening or diagnostic testing is recommended for men who are aged 45 years or more, but genetic counseling, routine ultrasound, and discussion of this phenomena are indicated.

If a disorder occurs as a result of a new mutation, the risk of recurrence in a sibling is generally low. However, an individual may be mosaic for an AD mutation, meaning that there are cells of more than one genetic constitution present. If germline mosaicism is present, an apparently unaffected individual will have an increased risk of having affected offspring. Germline mosaicism is thought to occur in approximately 16% of cases of the perinatal lethal disorder osteogenesis imperfecta type II and can also occur in X-linked disorders such as Duchenne muscular dystrophy and hemophilia A. This means that the rate of recurrence is increased and is about 1.3% to 4% with osteogenesis imperfecta.
Penetrance and Expression
Further complicating recurrence risk counseling and clinical evaluation of AD disorders are the phenomena of penetrance and variable expression. A given individual may have a disease genotype but may not express the phenotype if a disorder is not 100% penetrant, as is the case with retinoblastoma, which affects only about 90% of individuals who inherit the causative mutation. Age is also an important consideration because a disorder such as polycystic kidney disease is about 80% to 90% penetrant by 20 years of age and nearly 100% penetrant by 30 years of age, although only with careful imaging of the kidneys. A disorder may also demonstrate variable expression among affected individuals, even within a family. Targeted physical examinations, radiologic investigation, and other studies may be required to identify features of a disorder in at-risk family members; NF is a notable example. Essentially all individuals inheriting a mutation in the NF gene have some clinical symptoms, although they may range from only mild skin manifestations to multiple debilitating neurofibromas. Referral to a geneticist is indicated for those families identified with risk for heritable disorders.
Autosomal-Recessive Disorders
AR disorders are classically described as those for which two gene mutations must be present for the disorder to manifest. Such disorders occur most commonly in persons whose healthy parents each carry a mutation in the same recessive gene. The recurrence risk to such carrier parents is 25% in each pregnancy. Because the carrier frequency of a given condition is usually low, these disorders most commonly occur with no prior family history. Multiple affected individuals may be identified in a sibship but not typically in multiple generations unless there is consanguinity or the disorder is particularly common. Consanguinity increases the risk of having offspring with AR disorders because of the shared genetic material within a family. The increased risk to parents who are first cousins to have a child with a major genetic or congenital abnormality is about 6%, or twice the background rate.
Again, with advances in genetics, the exceptions to the traditional rules of AR inheritance are increasingly evident. It is now appreciated that carriers of many of these conditions may have subtle symptoms. Examples of this are cystic fibrosis (CF), in which male carriers have increased susceptibility to chronic pancreatitis and congenital bilateral absence of the vas deferens. Similarly, sickle cell carriers may suffer splenic infarct at high altitudes and are at increased risk for urinary tract infection in pregnancy.
AR disorders demonstrate much less variation in expression, and lack of penetrance is rarely encountered, so counseling is more straightforward with these conditions. Genetic heterogeneity due to more than one causative locus, or multiple alleles at a single locus, is the major cause of variation in severity of a single disorder.
X-Linked Inheritance
X-linked disorders can be dominant or recessive. Most X-linked conditions are recessive and are traditionally thought of as affecting only men, who have unaffected carrier mothers. Because women carry two X chromosomes whereas men carry only one, one X chromosome is randomly inactivated early in embryonic development, thereby allowing women to produce X-linked gene products in the same quantities as men (Lyon hypothesis). As X inactivation is random, some female heterozygote carriers of X-linked disorders will inactivate primarily their X chromosomes carrying the normal allele and therefore be symptomatic due to skewed X inactivation. This occurs in hemophilia A, in which some carriers can have a mild bleeding disorder because of reduced levels of factor VIII. Such affected women have symptoms that are usually, but not always, milder than their male counterparts. With isolated X-linked recessive disorders, it is helpful when possible to determine if an isolated case represents a new mutation or if the mother is a carrier. When molecular diagnosis or other methods of carrier detection are available, the situation can be clarified. However, such tests are not available for all conditions, and risks must often be empirically determined based on pedigree analysis and consideration of other factors such as the number of unaffected men in the pedigree. Increasingly, genomic sequencing is available to identify genetic mutations in individuals in whom a genetic disorder is suspected.
X-linked dominant disorders are less common than X-linked recessive diseases, and some such conditions are lethal in men. Examples include Rett syndrome, incontinenti pigmenti, and Aicardi syndrome. Aicardi syndrome is a rare disorder primarily of the CNS, including agenesis of the corpus callosum, microphthalmia, and infantile spasms. When an affected child with such a condition is born to healthy parents, it is likely due to a new mutation and the recurrence risk is low, although consideration must be given to germline mosaicism.
Fragile X syndrome, the most common inherited form of mental retardation, is an important disorder that demonstrates X-linked inheritance. Fragile X syndrome is characterized not only by mental retardation but also by behavioral difficulties including autism, and specific dysmorphic features. The condition affects males (approximately 1/4000) more commonly and more severely than females. Fragile X syndrome is a trinucleotide repeat disorder, characterized by expansion of DNA triplets beyond the normal, stable threshold, which may occur in certain genes and result in a condition or disease. Trinucleotide repeat disorders are typically neuromuscular diseases and include Huntington disease, myotonic dystrophy, Friedreich ataxia, and spinocerebellar ataxia. The number of repeats can expand due to chromosomal instability during meiosis, which can cause the phenotype of the condition to change and become more severe. Because of this, trinucleotide repeat disorders generally show genetic anticipation, in which the severity increases with each successive generation that inherits them. Individuals are considered to be carriers for fragile X syndrome when they have an intermediate number of trinucleotide (CGG) repeats in the FMR1 gene (also called a premutation) that can expand during oogenesis to a larger number of repeats (full mutation) (see Fig. 2-5 ). Full-mutation men have moderate to severe mental retardation and behavioral problems such as autism, whereas women are affected by a more variable phenotype, ranging from normal to as severe as affected men. Premutation female carriers have a relatively high rate of premature menopause (15% to 25%), whereas male premutation carriers can have a tremor ataxia condition with an onset later in life. For any patient with a family history of mental retardation, early menopause, or autism without an attributable diagnosis, or adults with parkinsonian features, carrier testing for fragile X syndrome is indicated.
Other Novel Genetic Mechanisms
Some genetic mutations have been noted to have a very different effect depending on the parent of origin, a phenomenon known as genetic imprinting. Imprinting is an epigenetic phenomenon by which certain genes are expressed or not expressed depending on the parent of origin ( Fig. 2-7 ). As depicted in Figure 2-8 , the imprinting cycle takes places in primordial germ cells early in development. A number of genetic disorders, including Prader-Willi syndrome (PWS) and Beckwith-Wiedemann syndrome (BWS), occur because of abnormalities of imprinted genes. Prader-Willi syndrome is characterized by neonatal and infantile hypotonia, hypogonadism, typical facial features, and feeding problems; these are followed in early childhood by excessive caloric intake resulting in morbid obesity and intellectual disability. The disorder occurs when there is no functional paternal copy of the SNRPN gene on chromosome 15. This can happen when both copies of chromosome 15 are maternally inherited, and no paternal copy is present, or when there is a deletion on the paternally inherited copy of chromosome 15. Several recent reports have indicated an association between imprinting disorders and in vitro fertilization (IVF) with intracytoplasmic sperm injection.


Mitochondria, which produce adenosine triphosphate (ATP), have their own unique DNA in addition to nuclear DNA. Mitochondria are all maternally inherited and have a high mutation rate. Many mitochondrial diseases have now been identified, and they are most commonly maternally inherited such that affected women pass the condition to all of their offspring and affected men do not pass the condition to any of their offspring. Mitochondrial disorders demonstrate variable expressivity within a family. Typical symptoms include neuromuscular abnormalities (such as loss of motor control, muscle weakness, and pain), gastrointestinal disorders and swallowing difficulties, poor growth, cardiac disease, liver disease, diabetes, respiratory complications, seizures, visual/hearing problems, lactic acidosis, developmental delays, and susceptibility to infection.
Multifactorial Inheritance
Many common congenital malformations, including NTDs, cleft lip and palate, and CHDs, have an increased risk of recurrence in families above the baseline population risk. A small percentage of such defects have a specific cause, such as single gene disorders, chromosomal abnormalities, or teratogens. Most, however, are isolated defects that result from complex interactions among a number of factors, including genotype at one or more loci, and a variety of environmental exposures that trigger, accelerate, or exacerbate the disorder. Therefore, occurrence does not match simple mendelian inheritance but is complex and multifactorial. Recurrence risk of such disorders is increased by the presence of more than one affected relative, a severe or early-onset form of disease, an affected person of less common sex (in disorders in which persons of one sex are more likely to be affected), high heritability of the disorder, and consanguineous parentage.
NTDs have long been thought to follow a typical complex inheritance pattern determined by multiple genetic and environmental factors. Known environmental risk factors include maternal obesity, diabetes, and medications such as valproic acid. The association between lower socioeconomic status and NTD led investigators to consider nutritional deficiencies as risk factors. Therefore, it was a remarkable discovery that folate supplementation reduced the risk for NTD by approximately 80%, although most women with a fetal NTD diagnosed during pregnancy have normal range folate levels. Public health initiatives to educate reproductive age women about the importance of folate and fortification of the food supply (cereals and breads) with folic acid have been instituted in the United States for the purpose of decreasing the incidence of NTDs. The mechanism for the protective action of folic acid in prevention of NTDs is not well understood. Recent data suggest folate deficiency is not problematic unless present in combination with a mutated gene or genes; together these risk factors result in the embryologic malformation.
Teratology
A milestone in human teratology occurred in 1941, when an Australian named Norman Gregg noted the association between first trimester maternal rubella infection and serious birth defects. Twenty years later, the thalidomide disaster sensitized the medical community to the risks of medications in pregnancy. Thalidomide, a very effective sedative, was widely used for several years in West Germany, Australia, and other countries until the association of prenatal exposure with phocomelia and amelia was recognized. Following the recognition of the potent teratogenicity of this drug, intense investigations into the safety of various medications in pregnancy heralded the modern age of teratology.
Embryos are more susceptible to teratogenic agents at specific critical periods of development ( Fig. 2-9 ). The preorganogenetic phase, from conception until somite formation, is known as the all-or-none period, when insults to the embryo are likely to result in either death of the conceptus and miscarriage or in intact survival without sequelae. At this stage, the embryo is undifferentiated, and repair and recovery are possible through multiplication of the still totipotential cells to replace those that have been lost. Exposure of embryos to teratogens during the presomitic stage usually does not cause congenital malformations unless the agent persists in the body beyond this period.
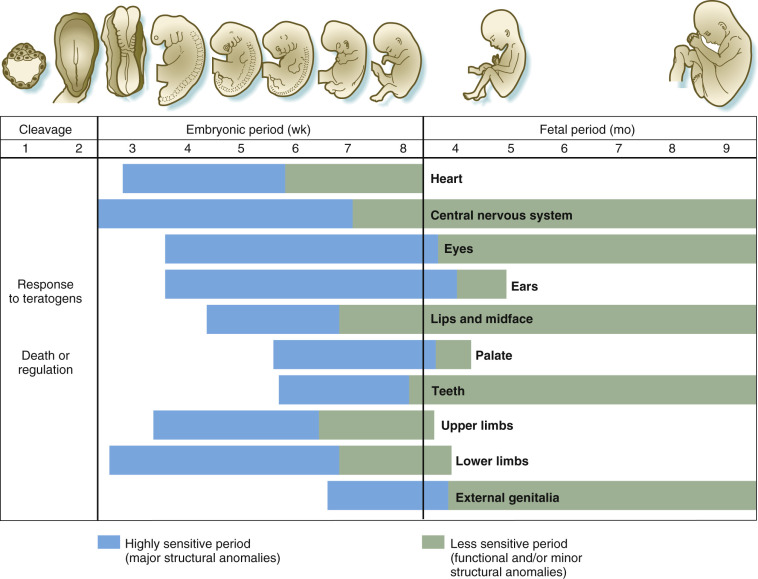
The embryonic period, between weeks 3 and 8 after conception, is when the basic steps of organogenesis occur. This is the period of maximum sensitivity to teratogenicity because tissues are differentiating rapidly and damage to them becomes irreparable. Exposure to teratogenic agents during this period has the greatest likelihood of causing a structural anomaly. Because teratogens are capable of affecting many organ systems, the pattern of anomalies produced depends upon which systems are differentiating at the time of teratogenic exposure. In general, organs that form first tend to be sensitive earlier, and very complex organs tend to have prolonged periods of high susceptibility to disruption.
The fetal phase, from the end of the embryonic stage to term, is the period when growth and functional maturation of organs and formed systems occur. Exposure to teratogens in this period will mainly affect fetal growth or the size or function of a specific organ. The brain in particular may be functionally affected almost throughout the entire pregnancy. Teratogens rarely cause gross structural anomalies during this time.
Recognized Teratogenic Agents
A large number of environmental factors have been reported to be associated with birth defects. They include chemical teratogens such as medications and hormones, maternal infections, and physical agents such as radiation. Although the list of medications suspected of teratogenicity is long, relatively few agents have been demonstrated to unquestionably cause birth defects in humans ( Table 2-4 ). In some cases these medications must still be used during pregnancy, as the benefit outweighs the risk they impose. Patients must be made aware of the risks and benefits of use in pregnancy to treat the maternal condition versus the potential for harmful fetal effects and maternal morbidity and mortality risks if untreated. Often, maternal uncontrolled diseases are more harmful to the fetus than the administered drug.
| Agent | Effects |
|---|---|
| ACE inhibitors | CNS defects, congenital heart defects (first trimester exposure) Renal failure, skull hypoplasia (second trimester exposure) |
| Alcohol | Fetal alcohol syndrome: intrauterine growth retardation, microcephaly, developmental delay, characteristic facies, CHD, cleft palate |
| Carbamazepine | Neural tube defects, similar features to fetal hydantoin syndrome |
| Chemotherapeutic agents | Miscarriage, various malformations |
| Cocaine | Abruptio placentae, prematurity, fetal loss, decreased birth weight, microcephaly, limb defects, urinary tract anomalies, and poor neurodevelopmental performance |
| Coumarin anticoagulants | Fetal warfarin syndrome: hypoplasia and calcific stippling of the epiphyses, intrauterine growth retardation, developmental delay, CNS damage, eye defects, hearing loss |
| Diethylstilbestrol | Clear cell adenocarcinoma, benign adenosis of the vagina, female genital structural changes (cervix/uterus) |
| Folic acid antagonists | Fetal aminopterin syndrome: CNS defects (hydrocephalus, meningomyelocele), facial anomalies (cleft palate, high-arched palate, micrognathia, ocular hypertelorism, external ear anomalies), abnormal cranial ossification, abnormalities of first branchial arch derivatives, intrauterine growth retardation, mental retardation |
| Hydantoin | Fetal hydantoin syndrome: craniofacial dysmorphology (wide anterior fontanel, ocular hypertelorism, metopic ridge, broad depressed nasal bridge, short anteverted nose, bowed upper lip, cleft lip, cleft palate), hypoplasia of the distal phalanges, nail hypoplasia, growth retardation, mental deficiency, cardiac defects |
| Isotretinoin | Retinoic acid embryopathy: craniofacial anomalies (microtia or anotia, accessory parietal sutures, narrow sloping forehead, micrognathia, flat nasal bridge, cleft lip and palate, and ocular hypertelorism), cardiac defects (primarily conotruncal malformations), abnormalities in thymic development, and alterations in CNS development |
| Lithium | Cardiac defects, especially Ebstein anomaly |
| Misoprostol | Limb defects, Moebius sequence |
| Organic mercury | Mental retardation, cerebral atrophy, spasticity, blindness |
| Sodium iodide (I-131) | Ablation of fetal thyroid gland |
| Thalidomide | Limb defects, including phocomelia, polydactyly, syndactyly, and oligodactyly; defects of the external ears; facial capillary hemangiomas; palsies of cranial nerve VI or VII; cardiovascular defects; agenesis of kidneys, spleen, gallbladder, and appendix; atresia or stenosis of esophagus, duodenum, and anus |
| Trimethadione | Fetal trimethadione syndrome: characteristic craniofacial anomalies, growth retardation, delayed psychomotor development, clefting, and congenital heart defects |
| Valproate | Neural tube defects, fetal valproate syndrome: narrow bifrontal diameter, high forehead, epicanthal folds, infraorbital creases, telecanthus, low nasal bridge, short nose with anteverted nares, midfacial hypoplasia, long philtrum, thin vermilion border, small mouth, cardiovascular defects, long fingers and toes, hyperconvex fingernails, and cleft lip |
| INTRAUTERINE INFECTIONS | |
| Cytomegalovirus | Cytomegalic inclusion disease: intrauterine growth retardation, microcephaly, chorioretinitis, seizures, blindness, optic atrophy Neonatal hepatosplenomegaly, jaundice, and thrombocytopenia |
| Rubella virus | Spontaneous abortion, congenital rubella syndrome: heart disease, deafness, cataracts, intrauterine growth retardation, encephalitis, abnormal long bones Neonatal hepatosplenomegaly, obstructive jaundice, thrombocytopenic purpura, mental retardation, neurologic deficits, and behavioral abnormalities |
| Treponema pallidum (causing syphilis) | Congenital malformations, prematurity, stillbirth, hepatosplenomegaly, osteochondritis or periostitis, jaundice, petechiae or purpuric skin lesions, lymphadenopathy, hydrops, edema, ascites, rhinitis, pneumonia, myocarditis, nephrosis, and bulbar pseudoparalysis |
| Toxoplasma (causing toxoplasmosis) | Encephalitis, hydrocephalus, intracranial calcifications, chorioretinitis, erythroblastosis, anemia, jaundice, hepatosplenomegaly, glomerulitis, myocarditis, myositis, CNS damage, seizures, mental retardation, cerebral palsy, deafness, and blindness |
| Varicella-zoster virus | Skin lesions, fetal growth retardation, limb hypoplasia, brain and eye defects, cerebral and cerebellar atrophy, seizures, developmental delay, and nerve palsies |
Maternal Infections
The incidence of intrauterine infections ranges between 3% and 15%. Rubella was one of the first infectious agents recognized to cause birth defects in humans and previously was one of the most common. Today, because of effective immunization programs against rubella, the number of affected fetuses has significantly decreased. Currently, the most common intrauterine infection with deleterious fetal effects is cytomegalovirus (CMV). Approximately 1 in 750 children born in the United States is born with or develops permanent problems as a result of this intrauterine infection. Features include hearing and vision loss, seizures, intellectual disability, and rarely death.
Intrauterine infections with long-term fetal sequelae may be viral, parasitic, or bacterial (see Table 2-4 ). The timing of maternal infection can influence both the severity and likelihood of fetal infection. For example, rubella causes a high percentage of malformations in the first trimester, whereas toxoplasmosis causes fetal infection more commonly in the third trimester but with more severe consequences if contracted earlier. Infection of the fetus may occur owing to hematogenous spread through the placental barrier or via ascending infection through the fetal membranes with direct contamination of the amniotic fluid.
Fetal CMV infection may be suspected by ultrasound features identified during routine surveillance or in the course of evaluation for a known maternal exposure. Ultrasound findings may include fetal ascites or nonimmune hydrops, ventricular dilation and periventricular calcifications, echogenic bowel (EB), calcifications within the fetal liver, fetal growth restriction (FGR), and placental thickening. Cases of suspected intrauterine CMV infections can be evaluated by measuring serum levels of maternal specific antibodies, but the significance of antibody levels is often uncertain. Moreover, a rise in levels of maternal antibodies is diagnostic for maternal, but not necessarily for fetal, infection. To diagnose fetal infection, the infective agent should be identified in the amniotic fluid by culture or polymerase chain reaction (PCR) or by specific immunoglobulin M (IgM) in fetal blood. At birth, viral culture of the neonatal urine or saliva is the gold standard.
Medications
Although many medications have been suspected of being human teratogens, there are a relatively small number for which there is convincing evidence linking the substance directly to congenital malformations in humans. Testing drugs to determine teratogenicity is difficult because agents may cause a high incidence of severe defects in animals but may not cause malformations in other species or in humans (e.g., cortisone causes cleft palate in mice but not humans). Conversely, thalidomide is a tragic example of an agent that is highly teratogenic in humans but not in animals. Antiepileptic drugs (AEDs) are classic examples of medications associated with structural birth defects as well as neurocognitive impairment. Risk for NTDs is increased with exposure to valproic acid (1-5% of exposed offspring) and carbamazepine (0.5-1.0%), but NTDs have not been reported with other AEDs. Valproate monotherapy in utero exposure has also been associated with significantly lower IQ scores at the age of 3 when compared to use of other AEDs. Therapy with more than one agent seems to compound the risk for not only structural changes but intellectual disability as well.
Radiation
Ionizing radiation is a potent teratogen, with the response dependent on the dose as well as the gestational age at which the embryo/fetus is irradiated. Experience with the Japanese atomic bomb survivors as well as women treated with therapeutic radiation in pregnancy provides evidence of the damaging effects of large doses of ionizing radiation in humans. In contrast, there is no evidence that radiation exposure at typical diagnostic levels (e.g., a chest radiograph with two views and fetal exposure of 0.02-0.07 mrad) poses a significant threat to the embryo, and noncancerous health effects are not evident for fetal doses of less than 5 rads. Estimates indicate a slight increase in leukemia (approximately 1.5-2 times over baseline risk) with exposures of 1 to 2 rads (risk increases from 1 in 3000 background to 1 in 2000 with exposure to 1-2 rads). Ionizing radiation at high doses (>5 rads) can cause a variety of anomalies in various organ systems. The most prominent radiation-associated abnormalities are defects in the CNS, ranging from NTD to neurocognitive impairment. For fetuses exposed between 8 and 15 weeks’ gestation, atomic bomb survivor data indicate that the decline in IQ score is approximately 25 to 31 points per 100 rads, at doses above 10 rads. At 16 to 25 weeks’ gestation, the average IQ loss is approximately 13 to 21 points per 100 rads, at doses above 70 rads. After 26 weeks, at doses above 100 rads, the risks for stillbirth and neonatal death increase. When evaluating the need for imaging or therapeutic radiation in pregnancy, the indication and both maternal and fetal health consequences should the exposure be rendered or withheld should be discussed with the woman.
Maternal Illnesses
A number of maternal illnesses and metabolic disorders have been implicated in the genesis of congenital malformations. Maternal diabetes is strongly associated with congenital malformations, with the risk to the embryo directly related to the degree of maternal glucose control. The most common abnormalities include CHDs and NTDs, whereas caudal regression is rare but of greatly increased frequency in infants of diabetic mothers. The phenotype of the infants of diabetic mothers also includes macrosomia, polyhydramnios, and stillbirth, with hypoglycemia and erythroblastosis (increased fetal nucleated red blood cells) common neonatal complications. Maternal prepregnancy obesity is independently associated with fetal malformations including cleft palate, CHDs, NTDs, and congenital diaphragmatic hernias. Untreated maternal phenylketonuria is also a potent teratogen and results in microcephaly, mental retardation, CHDs, and low birth weight. Risks to the fetus are related to the level of maternal phenylalanine and almost entirely eliminated with maternal dietary management and avoidance of phenylalanine in the diet of affected women during pregnancy.
Mechanical Factors
Mechanical disruption can result in a number of recognizable and characteristic sequence disorders. Mechanical forces may include multiple gestations, uterine myomas that distort the uterine cavity, or amniotic bands. Often, the underlying cause is less important than the common disruption pathway. Potter sequence, for example, results from early, longstanding oligohydramnios of any cause and consists of pulmonary hypoplasia, clubfeet, congenital hip dislocation, low-set ears, micrognathia, and flattened faces. This same phenotype can result from different causes of absent amniotic fluid, including bilateral renal agenesis and early rupture of the amniotic membrane.
Related posts:
 Ultrasound Evaluation of Fetal Aneuploidy in the First and Second Trimesters
Ultrasound Evaluation of Fetal Aneuploidy in the First and Second Trimesters
 Measurements Frequently Used to Estimate Gestational Age and Fetal Biometry
Measurements Frequently Used to Estimate Gestational Age and Fetal Biometry
 Role of Doppler Sonography in Obstetrics
Medications and Reported Associated Malformations
Role of Doppler Sonography in Obstetrics
Medications and Reported Associated Malformations
 Role of Sonography in Gynecologic Interventions
Role of Sonography in Gynecologic Interventions
 Ultrasound Features of Fetal Syndromes
Ultrasound Features of Fetal Syndromes
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


