Abstract
Holt-Oram syndrome, also known as hand-heart syndrome, is classically described to have upper limb anomalies affecting the thumb and heart, mostly septal defects. The syndrome may occur sporadically but commonly follows autosomal dominant transmission. Most cases are caused by a mutation in the transcription factor gene TBX5, located on 12q24.1. Holt-Oram has 100% penetrance and variable expressivity. If the disorder is suspected prenatally, thorough evaluation of the fetal heart is warranted because more than 75% of fetuses are affected with congenital heart disease. Similarly, due to the upper extremity involvement, the fetal radius-ulna-hand complex needs to be evaluated. A multidisciplinary approach is imperative, including a geneticist, hand surgeon, pediatric cardiologist, and maternal-fetal medicine subspecialist.
Keywords
Holt-Oram syndrome, thumb malformation, congenital heart disease, upper extremity malformations, cardiac arrhythmia, atrial and ventricular septal defects
Introduction
Holt-Oram, also known as hand-heart syndrome I, was first described in 1960 by Mary Clayton Holt and Samuel Oram as a constellation of malformations that they observed in four generations of one family. The original description included atrial septal defects and a thumb anomaly that lay in the same plane as the finger. Individuals affected by this disorder have the classically described upper limb anomaly and heart defects (mostly atrial septal defects), but may additionally present with pectus excavatum or carinatum and vertebral anomalies. Holt-Oram is now known to occur because of a mutation in the TBX5 gene located on 12q24.1 and is a rare autosomal dominant syndrome characterized by high penetrance and variable expression.
Disorder
Definition
Hallmark features of Holt-Oram syndrome include cardiovascular defects, most of which are atrial septal defects (up to 60%), and radial arm malformation affecting the thumb. Other possible cardiac anomalies include arrhythmias and complex cardiac malformations, most commonly atrioventricular canal defects and tetralogy of Fallot. Other potential syndromic features include variable upper limb anomalies and skeletal anomalies. This syndrome has been associated with over 70 unique mutations in the transcription factor TBX5 located on 12q24.1.
Prevalence and Epidemiology
Holt-Oram syndrome has a mean prevalence of 0.7 : 100,000 and is typically diagnosed prenatally or in the early years of life. There is high regional variation; in Ireland, the prevalence is 0.2 : 100,000 and in Belgium, 1.4 : 100,000. There appears to be a significant lower prevalence rate in more modern reports (post-2001) compared to earlier epidemiologic studies; however, this finding has no obvious explanation. The male-to-female ratio is close to 1 : 1.
Etiology, Pathophysiology, and Embryology
Holt-Oram presentation may be sporadic, but more commonly it follows autosomal dominant transmission with nearly 100% penetrance but variable expressivity. Individual carriers of TBX5 allelic variants have a high variability of clinical presentation even within families. Two groups, Li et al. and Basson et al. determined that Holt-Oram syndrome is caused by a mutation in TBX5. TBX5 mutations have been noted in 30%–75% of patients clinically suspected to have Holt-Oram syndrome. Pathogenic mutations in this gene are typically inactivating, such as deletions, nonsense, and truncations, though missense mutations in functional domains have also been described. TBX5 is located on the long arm of chromosome 12, locus 12q24.1. TBX5 is a member of the T-box family of transcription factors, and has been found to be critically important in cardiac and limb development. Pathogenic mutations typically affect the ability of the subsequent protein to dimerize with other proteins, with the end result being dysregulation of function.
Manifestations of Disease
Clinical Presentation
Cardiac disease when present may include dysrhythmias and/or malformations. The prevalence of congenital heart disease (CHD) in individuals affected with Holt-Oram syndrome is reportedly between 75% and 95%. The most common cardiac malformations are septal defects (54.2%) (atrial septal defect > ventricular septal defect), whereas 25% are considered complex CHD. Individuals should be followed with pediatric cardiology to time surgical intervention as there is a risk of development of pulmonary hypertension in uncorrected septal defects. In addition, affected individuals should be assessed for possible arrhythmias such as bradycardias and atrioventricular conduction abnormalities that may require placement of a pacemaker.
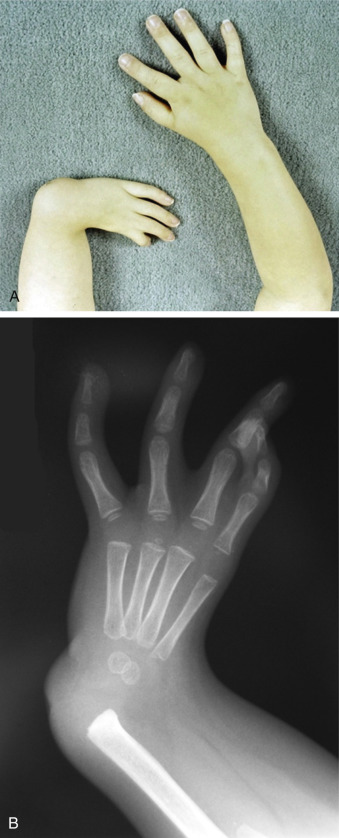
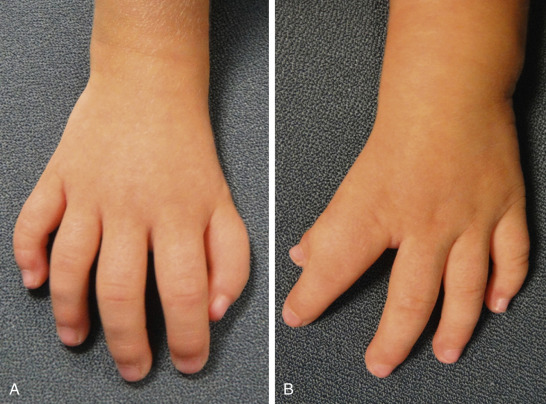
Skeletal abnormalities, particularly in the upper extremities, are the other main class of defect in Holt-Oram syndrome ( Fig. 132.1 ). The most commonly observed defects are agenesis or hypoplasia of the radius, aplasia or hypoplasia of the ulna, and phocomelia. Defects involving the thumb are exceptionally common ( Fig. 132.2 ). The defect can present as a missing thumb, or a thumb that appears as a finger. Skeletal abnormalities are usually bilateral in 84% of cases and can be similar in phenotype, but in cases where they differ, the left side is typically more severely affected than the right. Chest anomalies, such as pectus excavatum, narrow sloping shoulders, and vertebral anomalies have also been described but are less common. Importantly for the genetic counseling of families, there is no known intellectual impairment associated with Holt-Oram syndrome. While the classic constellation of malformations in conjunction with an autosomal dominant distribution in a family is sufficient for clinical diagnosis, often there is a lengthy differential diagnosis and genetic studies may be needed for confirmation.