The main role of computed axial tomography in the field of movement disorders was to exclude uncommon but potentially reversible structural abnormalities including tumors, chronic subdural hematoma, and communicating hydrocephalus presenting with parkinsonism. In the past 20 years magnetic resonance has had greater impact in facilitating accurate diagnosis but its clinical usefulness is less than in some other neurologic fields. Dopamine transporter SPECT imaging is helpful in distinguishing benign tremulous Parkinson disease from atypical tremor syndromes and other clinical scenarios where the demonstration of nigrostriatal dopamine denervation is helpful. We use eight case vignettes to illustrate how MR imaging findings can assist in the diagnosis of movement disorders and, in some cases, change the course of patient management.
The main role of computed axial tomography in the field of movement disorders was to exclude very uncommon but potentially reversible structural abnormalities including tumors, chronic subdural hematoma, and communicating hydrocephalus presenting with parkinsonism. In the past 20 years, magnetic resonance has had a far greater impact in facilitating accurate diagnosis but it would be fair to say that its clinical usefulness is much less than in some other neurologic fields such as cerebrovascular disease and multiple sclerosis. Dopamine transporter single-photon emission computed tomography (SPECT) imaging (DAT scan) on the other hand has proved to be a very helpful investigation in distinguishing benign tremulous Parkinson disease (PD) from atypical tremor syndromes and in a number of other clinical scenarios where the demonstration of nigrostriatal dopamine denervation is helpful.
In this article, we use eight case vignettes to illustrate how MR imaging findings can assist in the diagnosis of movement disorders and, in some cases, change the course of patient management.
Case 1: A 68-year-old man with tremor, ataxia, and parkinsonism
A 68-year-old man developed postural tremor of his right hand at the age of 60. Two years later, his gait became progressively unsteady. There was no family history of relevance. Examination revealed a mild dysexecutive syndrome, bilateral Holmes tremors, a wide-based ataxic gait and symmetric bradykinesia. There was no response to L-dopa and an MR scan was ordered.
Neuroimaging
This patient had Holmes tremor, cerebellar ataxia, parkinsonism, and subtle cognitive dysfunction. At initial presentation he was thought to have tremulous PD. Coexisting cerebellar ataxia should also lead to consideration of atypical parkinsonian syndrome such as the cerebellar subtype of multiple system atrophy (MSA-C) or other inherited cerebellar ataxias such as spinocerebellar ataxia (SCA) type 2 and 3.
The MR imaging scan revealed increased T2 signal in the white matter of middle cerebellar peduncles (the “MCP sign”) ( Fig. 1 ). This is a characteristic feature observed in Fragile X tremor ataxia syndrome (FXTAS). This is a recently recognized adult disorder that occurs in individuals who are carriers of premutation alleles (55–200 CGG repeats) of the Fragile X Mental Retardation (FMR1) gene, leading to production of toxic FMR1 mRNA and formation of ubiquitin-positive intranuclear inclusions in the neurons and astrocytes throughout the brain.
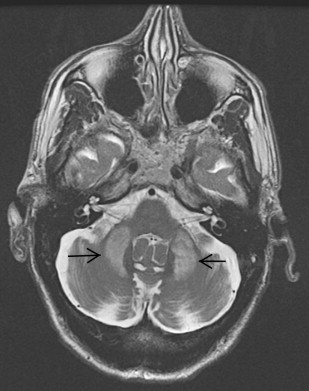
Prevalence of FXTAS is estimated at 1 in 8000 males older than 50 years in the general population and is still frequently unrecognized. Presenting symptoms include postural or intention tremor followed by gait ataxia. Other common features include L-dopa unresponsive parkinsonism, frontal lobe signs leading to cognitive dysfunction, dysautonomia, and peripheral neuropathy. The disorder is more slowly progressive than MSA-C. A study showed that the frequency of FMR1 premutation alleles was 4 (2.4%) in 167 patients who received a clinical diagnosis of probable MSA-C. Relevant family history includes children or grandchildren with mental retardation or behavioral disorders, female relatives with infertility, premature menopause, or family members with ataxia, psychiatric problems, dementia, or diagnosis of PD with dementia or multiple sclerosis.
The presence of the characteristic “MCP sign” with the finding of 87 CGG repeats (55–200) in the FMR1 gene led directly to the correct diagnosis in this case. The “MCP sign” is seen in 60% of males and 13% of females with the FXTAS permutation, so has limited sensitivity. Patients with both tremor and ataxia and who tested positive for the premutation but lack the “MCP sign” are diagnosed as having “probable” FXTAS. At autopsy, only a mild degree of spongiosis with occasional swollen axons are found in the MCP region despite the significantly abnormal appearance on MR imaging.
The “MCP sign” can also be rarely observed in other conditions such as MSA-C, SCA type 6, Wilson disease, acquired hepatocerebral degeneration, cerebrovascular infarction, and dysplastic changes in neurofibromatosis.
Other radiological findings observed in FXTAS include moderate to severe generalized brain atrophy and increased signal in T2-weighted images in periventricular and deep white matter of the cerebral hemispheres and corpus callosum.
Case 2: A 14-year-old teenager with progressive cognitive decline, dysarthria, dystonia, and spastic gait
A 14-year-old boy presented with a 2-year history of progressive dysarthria, dystonia of upper limbs, and gait difficulty. His test results from school had declined markedly over the past year and he had personality change with frequent violent outbursts. He was an only child and had no family history of neurologic disorder. Examination showed bradyphrenia, dysarthria with palilalia, moderate limb dystonia and rigidity pyramidal signs, and a spastic gait.
Neuroimaging
In a young patient presenting with a mixed movement disorder, pyramidal abnormalities, and cognitive decline, the diagnosis of Wilson disease should always be ruled out. Rare neurodegenerative and neurometabolic disorders such as neuronal ceroid lipofuscinosis (Kuf disease), Westphal variant of Huntington disease (HD), neurodegeneration with brain iron accumulation (Hallervorden-Spatz syndrome), hexosaminidase A, or GM1 galactosidase deficiency should also be considered.
In this case, the finding of an “eye of the tiger” sign on neuroimaging led to the correct diagnosis of neurodegeneration with brain iron accumulation (Hallervorden-Spatz syndrome) ( Fig. 2 ).
Neurodegeneration with brain iron accumulation (NBIA) is an autosomal recessive disorder characterized by dystonia, parkinsonism, and iron accumulation in the brain. Many patients with NBIA have mutations in the pantothenate kinase-2 gene (PANK-2) on chromosome 20p13. The characteristic “eye of the tiger sign,” with hyperintense signal changes in the central part of the globus pallidum representing iron deposition, is strongly correlated with PANK-2 mutation, and all PANK-2 mutation-positive patients are said to eventually exhibit this radiological finding. Despite a recent case report describing the disappearance of the “eye of the tiger” in a case with genetically confirmed PANK-2 mutation, the sensitivity of this sign still approaches 100%. The reverse is also true, in that the “eye of the tiger” pattern is not observed in the MR imaging of PANK-2-negative patients. Although NBIA with PANK-2 mutation is most commonly a disorder of childhood, there are some cases with juvenile or adult presentations with dystonia, parkinsonism, and pyramidal signs. Severe dysarthria is often a prominent early finding.
“Eye of the tiger” sign is not pathognomonic, as it has been reported in some parkinsonian syndromes including three patients with progressive supranuclear palsy (PSP), one of whom was pathologically proven, one patient with early-onset levodopa-responsive parkinsonism, and one patient with clinical diagnosis of corticobasal degeneration (CBD). The clinical picture and age of onset is different in these disorders and should not lead to diagnostic confusion. On the other hand, the “eye of the tiger” sign has recently been reported in two cases of neuroferritinopathy, which is another NBIA condition of autosomal dominant inheritance that can closely resemble neurodegeneration with brain iron accumulation with the PANK-2 mutation.
Case 2: A 14-year-old teenager with progressive cognitive decline, dysarthria, dystonia, and spastic gait
A 14-year-old boy presented with a 2-year history of progressive dysarthria, dystonia of upper limbs, and gait difficulty. His test results from school had declined markedly over the past year and he had personality change with frequent violent outbursts. He was an only child and had no family history of neurologic disorder. Examination showed bradyphrenia, dysarthria with palilalia, moderate limb dystonia and rigidity pyramidal signs, and a spastic gait.
Neuroimaging
In a young patient presenting with a mixed movement disorder, pyramidal abnormalities, and cognitive decline, the diagnosis of Wilson disease should always be ruled out. Rare neurodegenerative and neurometabolic disorders such as neuronal ceroid lipofuscinosis (Kuf disease), Westphal variant of Huntington disease (HD), neurodegeneration with brain iron accumulation (Hallervorden-Spatz syndrome), hexosaminidase A, or GM1 galactosidase deficiency should also be considered.
In this case, the finding of an “eye of the tiger” sign on neuroimaging led to the correct diagnosis of neurodegeneration with brain iron accumulation (Hallervorden-Spatz syndrome) ( Fig. 2 ).
Neurodegeneration with brain iron accumulation (NBIA) is an autosomal recessive disorder characterized by dystonia, parkinsonism, and iron accumulation in the brain. Many patients with NBIA have mutations in the pantothenate kinase-2 gene (PANK-2) on chromosome 20p13. The characteristic “eye of the tiger sign,” with hyperintense signal changes in the central part of the globus pallidum representing iron deposition, is strongly correlated with PANK-2 mutation, and all PANK-2 mutation-positive patients are said to eventually exhibit this radiological finding. Despite a recent case report describing the disappearance of the “eye of the tiger” in a case with genetically confirmed PANK-2 mutation, the sensitivity of this sign still approaches 100%. The reverse is also true, in that the “eye of the tiger” pattern is not observed in the MR imaging of PANK-2-negative patients. Although NBIA with PANK-2 mutation is most commonly a disorder of childhood, there are some cases with juvenile or adult presentations with dystonia, parkinsonism, and pyramidal signs. Severe dysarthria is often a prominent early finding.
“Eye of the tiger” sign is not pathognomonic, as it has been reported in some parkinsonian syndromes including three patients with progressive supranuclear palsy (PSP), one of whom was pathologically proven, one patient with early-onset levodopa-responsive parkinsonism, and one patient with clinical diagnosis of corticobasal degeneration (CBD). The clinical picture and age of onset is different in these disorders and should not lead to diagnostic confusion. On the other hand, the “eye of the tiger” sign has recently been reported in two cases of neuroferritinopathy, which is another NBIA condition of autosomal dominant inheritance that can closely resemble neurodegeneration with brain iron accumulation with the PANK-2 mutation.
Case 3: A 42-year-old man with progressive cognitive decline, orolingual dyskinesia, palatal tremor, and dystonia of the legs
A 42-year-old man developed stereotyped hyperkinetic movements of the tongue and lips and slurred speech at the age of 35. His father and paternal grandmother had both developed a similar movement disorder in middle age. Examination revealed impairment of abstract reasoning, a dysexecutive syndrome, and the presence of some frontal release signs. Eye movements were abnormal with hypometric saccades and the use of head thrusts to initiate saccades. He had a slurring dysarthria, orofacial dyskinesia, and involuntary pouting with occasional facial grimacing. Palatal tremor at a frequency of 1 Hz was noted but ear clicking was not a feature. Lower limb examination revealed both dystonia and chorea of the right leg and a striatal right toe.
The following test results were normal or negative: full blood count, peripheral blood smear, copper studies, creatine kinase (CK), liver function test, nerve conduction studies, and genetic testing for Huntington disease.
Neuroimaging
The clinical picture and family history suggestive of an autosomal disorder raised the possibility of Huntington disease. The prominence of orofacial dyskinesias and the presence of both chorea and dystonia also raised the possibility of neuroacanthocytosis and McLeod syndrome. However, the absence of peripheral neuropathy on nerve conduction studies and a normal CK effectively excluded these disorders. Genetic test for Huntington disease was negative, ruling out Huntington disease. Some of the autosomally inherited spinocerebellar ataxias and ataxia telangiectasia and dentatorubropallidoluysian atrophy (DRPLA) may present with a movement disorder and cognitive deficit but cerebellar ataxia is usually prominent. Genetic testing for SCA 1 to 3, 6, and 7 DRPLA was negative in this case and ataxia telangiectasia was also ruled out by normal alpha fetoprotein levels.
Hypointense signal on T2-weighted MR imaging in the globus pallidus observed in this case raised the possibility of a neurodegenerative disorder in which brain iron accumulation was prominent ( Fig. 3 ). This is a characteristic feature in pantothenate kinase-associated neurodegeneration (Hallervorden-Spatz disease), infantile neuroaxonal dystrophy (INAD) with PLA mutation, aceruloplasminemia, and neuroferritinopathy. The clinical picture and radiological findings in this case pointed more toward neuroferritinopathy or aceruloplasminemia, where more frequent involvements of the dentate and putamen are found radiologically and at postmortem and where the “eye of the tiger” sign is absent. Genetic testing confirmed the clinical suspicion of neuroferritinopathy in this case.
Neuroferritinopathy is an autosomal dominant condition that was first described in 2001 in families originating in the Cumbrian Lake District region of northwest England. A French family with neuroferritinopathy with the same mutation as the British family was later described. Patients have a single adenine insertion between nucleotides 460 and 461 in exon 4 of the ferritin light chain gene. Ferritin is an iron-storage protein and alteration of its structure by gene mutation leads to accumulation of extracellular iron in the brain, particularly in the basal ganglia. Excessive iron accumulation is toxic to the neurons, which leads to oxidative stress and neurodegeneration. The clinical suspicion of this disorder can be confirmed by the finding of serum ferritin being abnormally low or at the low end of the normal range. Definitive diagnosis is made by genetic testing confirming ferritin light chain gene mutation.
Patients present in their 20s to 40s with abnormal movements composed of dystonia, dysarthria, blepharospasm, chorea, spasticity, rigidity, and cognitive impairment of frontal subcortical dysfunction and sometimes the disease can progress rather acutely. These abnormal movements are characteristically asymmetric at initial presentation and remain asymmetrical despite gradual generalized involvement as the disease progresses. Predilection of the facial and buccolingual regions are common. Palatal tremor has been reported. It is thought to be caused by iron deposition in the dentate nuclei, which is a component of the “Mollaret triangle” in addition to the inferior olivary and red nuclei.
A confluent area of hyperintensity involving globus pallidus and putamen with a rim of peripheral hypointensity is characteristic on MR imaging. This finding corresponds to the autopsy finding of cystic degeneration of the putamen and globus pallidus with fluid accumulation within the cysts.
In patients with palatal tremor, the MR imaging finding of inferior olivary nucleus hypertrophy suggests “symptomatic palatal tremor,” and clinically, as in this case, is not associated with ear clicking. Symptomatic palatal tremor has also been reported in MSA, PSP, and late-onset Alexander disease where atrophy of the medulla oblongata in the presence of white matter lesions are helpful radiological findings.
Case 4: A 42-year-old drug addict with parkinsonism, early gait instability, and falls backward
A 42-year-old Ukrainian man presented with progressive slurred speech, asymmetrical akinetic rigidity, gait difficulty, and emotional lability. He had a history of intravenous drug abuse. Examination revealed incontinent laughter, risus sardonicus, hypomimia, apraxia of eyelid opening, hypophonia, asymmetrical bradykinesia, and rigidity. He fell backward spontaneously and had a characteristic “cock walk gait” with an erect posture. His eye movements were normal. The symptoms did not improve with L-dopa therapy.
Neuroimaging
This middle-aged man developed a progressive parkinsonian syndrome with several features that would be unusual for PD such as gait difficulty falls backward, retropulsion, pseudobulbar symptoms, apraxia of eyelid opening, and poor L-dopa response within the first 2 years of illness. These symptoms more closely resemble PSP with the exception of a “cock walk gait,” in which patients walk on their toes with elbows flexed and an erect trunk. The clinical picture also resembled chronic manganism seen in miners exposed to ore fumes. His history of drug abuse and his country of origin raised the possibility of “ephedrone” abuse. This new form of manganese poisoning has been reported in drug addicts in Russia, Ukraine, Georgia, and Estonia who intravenously inject themselves with a cocktail of pseudoephedrine (cold remedy bought over the counter), mixed with potassium permanganate and vinegar as oxidants to produce methcathinone, a central nervous stimulant structurally similar to methamphetamine.
The clinical syndrome of “ephedrone” toxicity is similar to the original reports of chronic manganism observed in Chilean miners following inhalation of ore dust. Other common causes of manganese poisoning can be seen in workers in alloy plant or dry battery factories, exposure to manganese-containing pesticides, prolonged total parenteral nutrition, and chronic liver disease with resulting impaired hepatic manganese metabolism.
In this case, the symmetric hyperintense T1-weighted signal in the globus pallidus was suggestive of deposition of paramagnetic substance such as manganese, iron, and copper ( Fig. 4 ). This characteristic MR imaging finding helped confirm the clinical suspicion manganese poisoning from “ephedrone” abuse. Increased signal on T1-weighted MR imaging can be caused by other conditions including accumulation of fat, melanin, calcifications, hemorrhages, and hypoxic conditions such as carbon monoxide poisoning, nonketotic hyperglycemia, and neurofibromatosis.
Related posts:
 Brain Magnetic Resonance Imaging Techniques in the Diagnosis of Parkinsonian Syndromes
Brain Magnetic Resonance Imaging Techniques in the Diagnosis of Parkinsonian Syndromes
 Current Role of Functional MRI in the Diagnosis of Movement Disorders
Current Role of Functional MRI in the Diagnosis of Movement Disorders
 Anatomy of the Substantia Nigra and Subthalamic Nucleus on MR Imaging
The Role of Imaging in the Diagnosis of Vascular Parkinsonism
The Role of Imaging in the Surgical Treatment of Movement Disorders
Anatomy of the Substantia Nigra and Subthalamic Nucleus on MR Imaging
The Role of Imaging in the Diagnosis of Vascular Parkinsonism
The Role of Imaging in the Surgical Treatment of Movement Disorders
 Role of Transcranial Ultrasound in the Diagnosis of Movement Disorders
Role of Transcranial Ultrasound in the Diagnosis of Movement Disorders
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


