, Marilyn J. Siegel2, Tomasz Miszalski-Jamka3, 4 and Robert Pelberg1
(1)
The Christ Hospital Heart and Vascular Center of Greater Cincinnati, The Lindner Center for Research and Education, Cincinnati, OH, USA
(2)
Mallinckrodt Institute of Radiology, Washington University School of Medicine, St. Louis, Missouri, USA
(3)
Department of Clinical Radiology and Imaging Diagnostics, 4th Military Hospital, Wrocław, Poland
(4)
Center for Diagnosis Prevention and Telemedicine, John Paul II Hospital, Kraków, Poland
Abstract
Hypertrophic cardiomyopathy (HCM) is a genetic cardiac disease characterized by diffuse or segmental left ventricle (LV) hypertrophy associated with a nondilated ventricular chamber, in the absence of other cardiac or systemic disease that might be capable of producing the same degree of hypertrophy [1]. It is an important cause of sudden cardiac death in any age group. The prevalence of HCM is approximately 0.2–0.5 % of the general population.
Hypertrophic cardiomyopathy (HCM) is a genetic cardiac disease characterized by diffuse or segmental left ventricle (LV) hypertrophy associated with a nondilated ventricular chamber, in the absence of other cardiac or systemic disease that might be capable of producing the same degree of hypertrophy [1]. It is an important cause of sudden cardiac death in any age group. The prevalence of HCM is approximately 0.2–0.5 % of the general population.
Pathologic hallmarks of HCM are myocyte disarray and interstitial fibrosis [2]. HCM is inherited as an autosomal dominant trait and is caused by mutations in a sarcomeric gene which encodes protein components of the cardiac sarcomeres.
HCM can be seen as part of certain genetic and metabolic syndromes. These include Noonan syndrome (associated with pulmonary valve dysplasia and septal defects), mitochondrial myopathies, glycogen storage diseases (type II Pompe disease), as well as other rare conditions. In older patients, other systemic diseases associated with left ventricular hypertrophy (LVH) include hypertension, Friedrich ataxia, pheochromocytoma, neurofibromatosis, tuberous sclerosis, amyloidosis, lentiginosis (referring to the presence of lentigines or spotted areas on the skin due to sun exposure), and Fabry disease, a rare X-linked autosomal recessive metabolic storage disorder caused by a lack of lysosomal a-galactosidase [3].
Imaging plays a role in detecting HCM, characterizing its pattern (phenotype), classifying disease severity, and providing risk stratification criteria for sudden cardiac death. Echocardiography is the conventional imaging modality to screen for this condition, as it can characterize the pattern and distribution of increased LV wall thickness. However, it can underestimate the degree of LV hypertrophy, especially at the apical endocardial border.
Cardiac MRI using delayed gadolinium-enhancement technique provides information regarding the location and extent of fibrosis and is considered the examination of choice to establish the diagnosis of HCM when echocardiography is inconclusive. It allows multiplanar imaging which has the advantages of allowing complete coverage of the myocardium and better characterization of the pattern and distribution of LV hypertrophy.
Cardiac CT is not routinely used in patients with HCM since it is associated with radiation exposure. However, CT can be useful in those patients in whom detailed anatomy of the coronary arteries is needed and in patients with contraindications to MRI such as a pacemaker or aneurysm clips. The spatial resolution of CT is usually superior to MRI, allowing exquisite, noninvasive coronary artery angiography. Cardiac CT also provides comprehensive anatomical and functional information about the heart chambers and myocardium [4]. In retrospectively gated cardiac CT acquisition, systolic anterior motion (SAM) of the mitral valve can be easily assessed. In addition, delayed enhancement CT scanning provides information about myocardial scarring and fibrosis similar to that provided by MRI [5, 6]. CT has been reported to be useful in the assessment of patients undergoing septal alcohol ablation by enabling depiction of the exact extent and location of septal hypertrophy, systolic anterior motion of the mitral valve, and mitral valve leaflet–septal contact as well as septal perforator anatomy, which is of utmost importance in guiding septal ablation [7, 8]. See Table 10.1 for the utility of CT in the evaluation of HCM.
Table 10.1
The utility of CT in the evaluation of HCM
Distribution of hypertrophy |
LV maximal wall thickness |
LV outflow tract obstruction |
Presence of fibrosis (delayed enhancement on CT) |
Presence of systolic anterior motion of the mitral valve |
Coronary anatomy |
Evaluation of septal perforator arteries before alcohol septal ablation |
It should be noted that myocardial bridging is common in patients with apical hypertrophy and has been reported in the majority of patients presenting with typical or atypical chest pain [9]. It can lead to myocardial ischemia by narrowing the intramyocardial segment of the affected coronary artery, which can be especially well seen on CT.
10.1 Phenotype Analysis
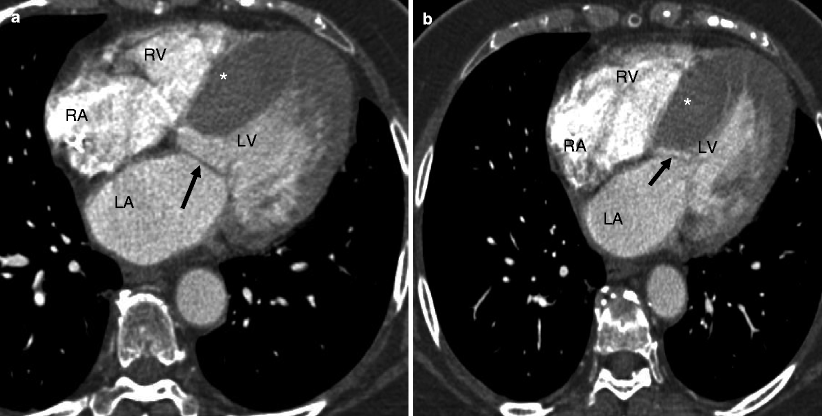
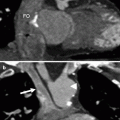
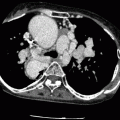
HCM is diagnosed when the LV wall thickness (typically the septum) is ≥15 mm in the end-diastolic phase [10]. It can be classified based on the distribution of the wall hypertrophy into the following phenotypes: asymmetric (Fig. 10.1) with and without dynamic LV outflow tract (LVOT) obstruction, apical (Fig. 10.2), midventricular (Fig. 10.3), and symmetric, mass-like, and noncontiguous [9] (Figs. 10.4a and 10.5) (see Table 10.2). HCM does not involve the basal or inferolateral segments of the LV, a helpful feature in differentiating HCM from other causes of LV hypertrophy [11]. Occasionally apical infarct can be identified by CT in the absence of coronary artery disease as a result of constriction of a distal LAD bridge (Fig. 10.6, black arrow). Recently, left ventricular myocardial crypts have been identified as a distinctive morphologic expression of HCM, occurring in patients with and without hypertrophy. Crypts may identify individual HCM family members before development of typical phenotype. Crypts are confined to the basal LV, most commonly in the ventricular septum or posterior LV free wall [12]. Example of LV crypt is presented in Fig. 10.6 (white arrow).
Get Clinical Tree app for offline access
Fig. 10.1
Representation of the asymmetric (septal) phenotype of HCM: panel (a) is an axial CT reconstructed in diastole. Panel (b




Related posts:

Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
