- •
Size and function of the left ventricle in comparison with the right ventricle.
- •
Direction of shunting at the atrial level (hypoplastic left heart syndrome is defined by inadequacy of the left-sided structures; hence, shunting at the atrial level in this anomaly is left to right).
- •
Presence and size of the mitral valve.
- •
Size of the aorta.
- •
Direction of shunting in the transverse aorta (retrograde in cases of aortic atresia or severe aortic stenosis).
- •
In cases of a patent mitral valve, the presence of coronary-cameral fistulae on color Doppler imaging of the small left ventricle.
- •
Anatomy of the atrial septum and patency of the foramen ovale.
- •
Pulmonary venous Doppler flow pattern as a reflection of adequate left atrial egress and an indirect gauge of possible restriction at the level of the atrial septum.
- •
Tricuspid valve function.
- •
Right ventricle function.
Anatomy and Anatomical Associations
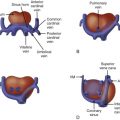
Hypoplastic left heart syndrome (HLHS) is not a single specific anomaly, but rather, a term that refers to a constellation of congenital cardiac anomalies in which the left side of the heart is insufficient in makeup and/or size to perform the task of providing for perfusion of the systemic circulation. The left ventricle (LV) may be nonviable as a provider of systemic perfusion owing to a spectrum of structural cardiac anomalies ranging from complete atresia of the mitral valve and aortic valves with no visible LV at all ( Figure 22-1 ) to hypoplasia of the mitral valve and aortic valves with borderline small LV cavity ( Figure 22-2 ). Hypoplasia of the ascending aorta, aortic arch, and coarctation of the aorta are commonly seen as well.


Noncardiac anomalies including chromosomal abnormalities are seen in 28% of infants with HLHS on autopsy study. Chromosomal abnormalities are seen in approximately 5% to 10% and include trisomy 13, trisomy 18, and Turner’s syndrome (monosomy X). One theory proposes a possible cause-and-effect relationship between the presence of large cystic hygroma as seen in Turner’s syndrome and compression of the developing left-sided structures of the heart, in particular the aortic arch, resulting in left ventricular hypoplasia. Rarely, deletion of chromosome 22q11 can occur. Syndromes such as Smith-Lemli-Opitz, Holt-Oram, and the VACTERL (vertebral abnormalities, anal atresia, cardiac abnormalities, tracheoesophageal fistula and/or esophageal atresia, renal agenesis and dysplasia, and limb defects) association have been reported.
Identification of deficits in neurocognitive outcome of some children with HLHS after surgical palliation has prompted a more rigorous search for neuroanatomical differences in the infant brain with HLHS from those with a normal heart. Subtle forms of cerebral dysgenesis such as an open cerebral operculum, micrencephaly (brain weight < 2 standard deviations below mean for age), and absence of the corpus collosum have been identified in newborns with HLHS at autopsy. The precise relationship between these anatomical features and late neurocognitive outcome has not yet been established. Infants with HLHS are commonly small for gestational age, with data emerging that suggest the presence of microcephaly for gestational age as well.
Frequency, Genetics, and Development
HLHS occurs in 0.26 per 1000 live births. The precise incidence varies somewhat because the definition of the syndrome and the time period of analysis differ slightly from study to study. Of all the various forms of congenital heart disease, HLHS is one of the most commonly diagnosed before birth because it can be easily detected by simple four-chamber view of the heart with absence of visualization of two normal-sized ventricles. Of 385 fetuses examined prenatally at The Children’s Hospital of Philadelphia (CHOP) with complex congenital heart disease requiring intervention between July 2002 and June 2005, 117 (30%) carried the diagnosis of HLHS. The recurrence risk for having another child with HLHS is relatively high in comparison with other forms of congenital heart disease and is 2% to 3%. The risk of a family with one child with HLHS having another person in the family with any form of congenital heart disease is also relatively high at 13% to 14%.
The developmental formation of HLHS is uncertain. There are no good animal models of HLHS, which has hampered our understandings of the origins of this anomaly. A number of theories exist. One is that poor development of the LV is a consequence of blood flow being diverted away from the left side of the heart early in development. The finding of abnormal atrial septal anatomy and restriction to flow found in so many patients suggests that perhaps limitation to right-to-left shunting, the primary source for filling of the LV in utero, may be the causative factor. With limited right-to-left shunting, there is less blood flow across the mitral and aortic valves; hence, the development of mitral, left ventricular, and aortic hypoplasia. Conversely, abnormal formation of the atrial septum may be secondary to an inability of the LV to fill for some other reason, perhaps altered compliance. Aortic stenosis may be the primary anomaly in some, with altered LV compliance and endocardial fibroelastosis limiting the filling capacity, resulting in poor growth. Alternatively, another theory holds to the view that none of these abnormalities are flow-derived one from the other, but rather, a series of associated, gene-signaled defects that occur simultaneously. The frequent occurrence of left- and right-sided valve dysplasia in HLHS probands and the increased prevalence of balloon aortic valvuloplasty in family members have influenced some investigators to conclude that HLHS is a severe form of valve malformation.
Based on the heterogeneity of anatomy seen in the HLHS, it seems likely that there are multiple pathways to achieve the diagnosis of a nonviable LV. Our definition of HLHS is, to a degree, a convenience and an arbitrary one, derived from the physiological consequences of the anomaly and our management. HLHS, as we define it today, is most likely an umbrella “grab-bag” term for a number of differently originating, but related, disorders.
Prenatal Physiology and Management
The intracardiac pathways of blood flow are different from normal in the fetus with HLHS. Superior and inferior vena caval return is to the right atrium and the ductus venosus drains in the normal fashion into the intrahepatic portion of the inferior vena cava. The eustachian valve, a membrane-like tissue at the orifice of the inferior vena cava that usually guides inferior caval flow toward the atrial septum, can at times be small and poorly formed. Pulmonary venous return is typically to the left atrium. However, as opposed to the normal right-to-left flow across the atrial septum, shunting occurs in a left-to-right manner across the atrial septum in order to decompress the left atrium when the LV is hypoplastic.
A number of anatomical features of HLHS affect the physiology and are worthy of discussion.
The Atrial Septum and Pulmonary Vasculature
Patency of the atrial septum through either a foramen ovale or an atrial septal defect is a critical factor in the normal development of the fetus with HLHS. A severely restrictive or intact atrial septum is seen in approximately 6% of infants with HLHS. In such cases, egress from the left atrium is obstructed, left atrial pressure is increased, and pulmonary venous return is impaired. Infants born with HLHS and an intact atrial septum are at high risk for mortality after birth. The fetus with an intact atrial septum is clinically asymptomatic because the placenta, and not the lung, is the organ of fetal oxygenation. However, at birth, these infants are extremely cyanosed and hypoxemic owing to the inability for oxygenated blood returning from the lungs to exit the left atrium and mix with the systemic venous return on the right side of the heart ( Figure 22-3 ). These infants do extremely poorly at birth with marked hypoxemia and early demise, despite prenatal diagnosis. Early postnatal identification and immediate intervention in infants with an intact atrial septum does not appear to substantially improve upon the relatively poor outcome in all, because it is believed that the inability for blood to exit the left atrium causes damage to the developing fetal lung vasculature in some fetuses. Histological analysis of the pulmonary veins in the newborn with HLHS and intact atrial septum reveals dilated vessels with thickened “arterialized” walls and multiple elastic laminae, suggesting a problem with pulmonary vascular development during fetal life ( Figure 22-4 ). Fetal intervention with balloon atrial septoplasty of the atrial septum has been performed and is in the early stages of development as a possible treatment for this disorder. The goal is to relieve left atrial hypertension and promote flow through the pulmonary veins by allowing for unimpeded egress out of the left atrium. The efficacy of the technique and the optimal timing of when in gestation to intervene in hopes of preventing the pulmonary vasculopathy that develops are yet to be determined. Ideally, it would be most helpful if such an intervention could take place before any permanent changes in pulmonary vasculature with vasculopathy takes place.


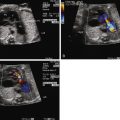
Identifying the precise nature of the patency of the atrial septum can be difficult. In addition, decompressing vessels such as a levoatrial cardinal vein may drain blood away from the left atrium, but yet not be detectable on fetal echocardiography. Hence, in addition to visualization of the septum, Doppler sampling in the pulmonary veins is of great use in gauging the presence and degree of impediment to left atrial egress. If there is an open, or only mildly restrictive, interatrial communication, there will be a normal pulmonary vein Doppler flow pattern consisting of a predominance of antegrade, biphasic flow into the atrium, with a short interval of low-velocity retrograde waveform concordant with atrial systole. As the degree of restriction at the atrial septum increases, the time interval and velocity of the retrograde atrial waveform will increase ( Figure 22-5 ). Investigators have determined that a velocity-time integral (area under the Doppler waveform) ratio of antegrade flow to retrograde flow of less than 5 suggests the presence of important atrial level restriction that may require attention and urgent atrial opening at birth.

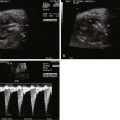
In addition to pulmonary vein flow analysis, we have added another component of direct evaluation of pulmonary vascular health in our fetuses with HLHS. In congenital diaphragmatic hernia, it is known that normal, healthy fetal pulmonary vasculature will vasodilate in response to maternal hyperoxygenation, whereas abnormal pulmonary vasculature will not. We, therefore, applied this principle to our mothers carrying fetuses with HLHS. By offering 100% oxygen through a non-rebreather mask for 15 minutes, adequate levels of oxygen should cross the placenta and result in relaxation of the pulmonary vasculature. We assessed the pulsatility index (PI) as a measure of vascular resistance and sampled at different sites in the pulmonary artery of fetuses with HLHS and varying degrees of atrial level restriction. The Doppler signals obtained from sampling in the pulmonary artery vary slightly from proximal to distal sites. We, therefore, sampled at a proximal pulmonary artery site just at the bifurcation (P1), midportion of the pulmonary artery as it just enters the lung parenchyma (P2), and distal in the lung bed beyond the hilum and into the lung parenchyma (P3). Case 22-1E and F demonstrates the contour change in the pulmonary artery Doppler waveform that takes place from room air sampling to maternal hyperoxygenation. There is a characteristic increase in peak velocity and an overall increase in diastolic flow and decrease in PI with maternal hyperoxygenation if the vasculature is normal. We found that fetuses with HLHS and an open atrial septum had good vasoreactivity, with at least a 10% decrease in PI with hyperoxygenation, whereas those with HLHS and an intact or highly restrictive septum had very little vasoreactivity and little change in PI with hyperoxygenation. The highest resistance measure is seen at site P3, the most distal aspect of the pulmonary artery measured. This is also the site of greatest sensitivity to change and vasodilation when the atrial septum is wide open. Those fetuses with little vasoreactive response to maternal hyperoxygenation were also those who had severe hypoxemia at birth, requiring urgent care and had in general poorer outcome. We advocate the use of maternal hyperoxygenation in all of our fetuses with HLHS and use the data to determine candidacy for either fetal atrial septoplasty intervention or urgent immediate opening of the septum at birth. Maternal hyperoxygenation is safe, with no untoward effects noted in the fetus and no evidence for ductal constriction in close to 100 fetuses thus far evaluated in this manner.
Patterns of Aortic Blood Flow
The pattern of blood flow in the aorta is altered in HLHS. In the absence of an LV or in aortic atresia, perfusion of the ascending aorta and, hence, the coronary circulation is in a retrograde manner originating distally, beyond the isthmus of the aorta, from the ductus arteriosus ( Figure 22-6 ). Often, in the presence of a small LV with aortic stenosis, a small amount of antegrade flow may take place in the ascending aorta; however, the transverse aorta and the cerebral vasculature arising therefrom are perfused retrograde from the ductus arteriosus. Dependency of the cerebral circulation upon retrograde perfusion from the ductus arteriosus into a variably hypoplastic aorta may limit the volume of blood flow and alter the regulatory mechanisms for cerebrovascular resistance. We have found lower cerebrovascular resistance as assessed by lower middle cerebral artery PI in fetuses with HLHS in comparison with normal and other forms of congenital heart disease. One explanation may be that the brain is autoregulating blood flow and attempting to increase perfusion by vasodilation under conditions in which there are structural impediments to flow, such as in HLHS.

Alterations in cerebral blood flow patterns in utero may be one explanation for the relatively high prevalence of neuroanatomical differences in infants with HLHS and may in part explain some of the neurocognitive deficits seen in a growing number of the survivors of surgery.
The Right Ventricle and Cardiac Output
Inadequacy of the LV to support the systemic perfusion has little obvious clinical impact in the fetus because the right ventricle takes over the task of blood flow delivery to the fetal body and placenta through ejection into the ductus arteriosus. In the absence of an adequate-sized LV, the right ventricle receives the full complement of venous return. Owing to the increase in blood volume, the right ventricle dilates, as does the tricuspid valve annulus. Ostensibly, on the surface of it, fetal perfusion looks to be preserved in the absence of right ventricular dysfunction or tricuspid valve insufficiency. However, upon deeper scrutiny, we have identified a decrease in the Doppler-derived measure of cardiac output in fetuses with HLHS with good ventricular function and no valvar dysfunction. Comparison of age-matched fetuses with HLHS and normal (mean gestational age 29 wk) reveals a lower “combined ventricular” cardiac output of 380 ± 124 mL/min/kg for the HLHS group versus 461 ± 45 mL/min/kg for the normal heart group ( P < 0.05). This is nearly a 20% decrease in cardiac output of the fetus with HLHS relative to normal. The overall impact of a diminished cardiac output for the developing fetus with HLHS is unclear. The vast majority of fetuses with HLHS progress to term without any difficulties. Hydrops fetalis of a cardiac etiology does not occur in HLHS unless there are problems with severe tricuspid regurgitation or severe right ventricle dysfunction. However, it is possible that a chronic low cardiac output may influence organogenesis and may contribute to the small weight for gestational age seen in HLHS. It may also have implications for mode of delivery because the fetus with a borderline poor cardiac output may not tolerate a long and strenuous vaginal delivery as well as a normal fetus. This area is ripe for further investigational work.
Prenatal Diagnostic Determination of Left Ventricle Inadequacy: Will the Left Ventricle Be Viable after Birth?
It is relatively straightforward to determine the inadequacy of the left-sided structures in the presence of mitral or aortic atresia with an absent or very small LV. However, cases in which there is ventricular size discrepancy between the LV and the right ventricle with a small borderline-sized LV can present a diagnostic challenge; it can be difficult to predict the viability and adequacy of the LV to provide for postnatal systemic perfusion. A number of abnormalities may result in the appearance of size discrepancy between the two ventricles ( Box 22-1 ). Distal aortic obstruction such as in coarctation of the aorta may present with a borderline small LV as may a number of anomalies in which the filling volume of the LV is altered from the normal. For example, in the presence of an interrupted inferior vena cava with azygos continuation to the superior vena cava, there is a decrease in the normal complement of streaming blood flow across the atrial septum. Filling of the LV is diminished, while an increased amount of flow is channeled to the superior vena cava and directed across the tricuspid valve, resulting in a relative increase in volume load to the right ventricle. Anomalous return of the pulmonary veins, either complete or partial, may limit volume filling of the LV in utero and provide for the appearance of a small cavity size. In congenital diaphragmatic hernia, abdominal contents may be positioned in the chest, potentially compressing the left-sided heart structures, thereby limiting LV filling by impeding right-to-left flow at the atrial level. In addition, lung compression and pulmonary hypoplasia may reduce the amount of pulmonary venous return to the LV, also contributing to a relatively smaller LV cavity volume.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


