This article reviews a variety of congenital and developmental disorders of the pediatric orbit with particular emphasis on ocular lesions, followed by a description of developmental and neoplastic orbital and ocular masses. The relationship of these diseases to various syndromes and/or known genetic mutations is also highlighted.
Key points
- •
Orbital diseases in children differ from those found in adults in terms of histopathologic and imaging characteristics.
- •
Clinical signs are often nonspecific, and imaging is a critical step in evaluating the pediatric orbit, optic pathway, and cranial nerves that supply the orbital contents.
- •
High-resolution 3-T MR imaging helps characterize orbital and ocular soft-tissue lesions, permitting superior delineation of orbital soft tissues, cranial nerves, blood vessels, and blood flow and detection of intracranial extension of orbital disease.
- •
Computed tomography (CT) is reserved primarily for evaluation of orbital bony architecture.
Introduction
The wide spectrum of orbital disease seen in children differs substantially from that found in adults in terms of histopathologic and imaging features. Clinical symptoms and signs such as proptosis, strabismus, diplopia, and optic disc edema are nonspecific, and diagnostic imaging studies play an essential role in depicting the nature and extent of orbital abnormalities, often providing a definitive diagnosis or a relevant differential diagnosis. The information provided by imaging is also important in determining optimal medical or surgical treatment and assessing response to treatment. In this article, the salient clinical and imaging features of various pediatric orbital lesions are described, and the differential diagnoses are reviewed.
Introduction
The wide spectrum of orbital disease seen in children differs substantially from that found in adults in terms of histopathologic and imaging features. Clinical symptoms and signs such as proptosis, strabismus, diplopia, and optic disc edema are nonspecific, and diagnostic imaging studies play an essential role in depicting the nature and extent of orbital abnormalities, often providing a definitive diagnosis or a relevant differential diagnosis. The information provided by imaging is also important in determining optimal medical or surgical treatment and assessing response to treatment. In this article, the salient clinical and imaging features of various pediatric orbital lesions are described, and the differential diagnoses are reviewed.
Normal anatomy
The orbital contents are contained within a bony pyramid. The orbital roof is formed by the orbital plate of the frontal bone. The lateral wall is formed by the orbital surface of the zygomatic bone and greater wing of the sphenoid. The frontal process of the maxillary bone, the lacrimal bone, lamina papyracea of the ethmoid bone, and the lesser wing of the sphenoid make up the medial wall from anterior to posterior. The orbital floor is formed by the orbital surfaces of the zygomatic, maxillary, and palatine bones. The optic foramen forms the apex of the bony pyramid and is formed by the lesser wing of the sphenoid. The superior orbital fissure is limited by the lesser wing of the sphenoid superomedially and the greater wing of the sphenoid inferolaterally. The inferior orbital fissure lies between the orbital floor and the greater wing of the sphenoid. The optic canal and superior and inferior orbital fissures transmit nerves and vessels ( Table 1 ); spread of tumor along these conduits can occur from the orbit to extraorbital compartments including intracranial extension.
| Foramen | Contents |
|---|---|
| Superior orbital fissure | Cranial nerves III, IV, V 1, VI Superior ophthalmic vein Orbital branch of middle meningeal artery |
| Inferior orbital fissure | Cranial nerve V 2 Infraorbital vein Infraorbital artery |
| Optic canal | Cranial nerve II Ophthalmic artery |
The orbital contents are divided into the intraocular compartment or globe, the muscle cone, and the intraconal and extraconal spaces ( Fig. 1 A). The extraocular muscles include the superior, inferior, medial, and lateral rectus muscles and the superior and inferior obliques; all but the inferior oblique muscles constitute the muscle cone (see Fig. 1 B). The levator palpebrae superioris lies superior to the superior oblique muscle. The extraocular muscles converge at the orbital apex to form a fibrous connective tissue ring known as the annulus of Zinn. The nonocular compartment of the eye is divided by the muscle cone into conal (muscle cone and annulus of Zinn), intraconal, and extraconal spaces. The intraconal space contains fat, the ciliary ganglion, the ophthalmic artery and vein, and branches of the ophthalmic nerve. The ophthalmic artery and vein and cranial nerves enter the intraconal space through the annulus of Zinn. The extraconal space contains fat, the lacrimal gland, and cranial nerves (branches of the ophthalmic and trochlear nerves). The superior oblique muscles receive motor supply from the trochlear nerves (cranial nerve IV). The lateral rectus muscles are innervated by the abducens nerves (cranial nerve VI), and the oculomotor nerves (cranial nerve III) supply motor function to the remaining extraocular muscles.
The globe consists of 3 distinct layers from the outside to inside: sclera, uvea, and retina (see Fig. 1 C, D). The choroid and retina are inseparable on routine cross-sectional imaging but can be differentiated in the presence of choroidal or retinal detachments. The uvea consists of the iris, ciliary body, and choroid (the most vascular structure of the globe). The retina continues posteriorly as the optic nerve. The collagenous sclera is continuous anteriorly with the cornea and posteriorly with the dura and appears hypointense on T1-weighted MR images at 3T (see Fig. 1 C).
Imaging technique
Imaging of the orbit is primarily accomplished by ultrasonography (US) (evaluation of the globe), CT (bony anatomy), and MR imaging (soft-tissue characterization). CT is indicated for the bony assessment in craniofacial anomalies, trauma, orbital complications of acute sinusitis (with contrast), and assessment of bony remodeling or destruction from orbital masses. Helical 2.5- to 3-mm axial images are obtained with multiplanar soft-tissue and submillimeter bone reformats. CT angiography (CTA) may be obtained for diagnosis or follow-up of suspected orbital arteriovenous malformation (AVM) or arteriovenous fistula (AVF). The parameters for CT should use the lowest dose possible while still providing diagnostic quality images.
Orbital MR imaging is optimally achieved at 3T using a 32-channel phased-array head coil or equivalent coil when possible. In some instances specialized orbital surface coils may be used. Imaging protocols depend on clinical indications. For example, suspected tumors are imaged with high-resolution, thin-section (<3 mm) axial and coronal fat-suppressed T2; axial non-echo planar diffusion-weighted imaging (DWI); axial T1 and multiplanar high-resolution, fat-suppressed, gadolinium-enhanced T1-weighted images. MR venography (MRV) and/or MR angiography (MRA) are sometimes indicated for vascular assessment. Heavily T2-weighted 3-dimensional sequences with submillimeter-thick images (eg, sampling perfection with application optimized contrasts using different flip angle evolution [SPACE], constructive interference in steady state [CISS], fast imaging employing steady state acquisition [FIESTA], sensitivity encoding [SENSE]) are of use for ocular assessment, especially for intraocular tumors such as retinoblastoma and for assessment of cranial nerves. Congenital strabismus and eye movement disorders generally require a combination of thin-section high-resolution axial and coronal T1-weighted images for assessment of the size, shape, and position of the extraocular muscles and imaging of the brain and relevant cranial nerves.
Conventional catheter angiography is reserved for the delineation of orbital AVM or AVF and sometimes for endovascular treatment.
Congenital and developmental anomalies
Anophthalmos and Microphthalmos
Anophthalmos or anophthalmia refers to congenital absence of the eyes. Anophthalmos and microphthalmos are a significant cause of congenital blindness and can be isolated or syndromic. Several genetic mutations involving PAX6, SOX2 , and RAX genes are associated with these conditions.
Primary anophthalmos is bilateral in approximately 75% of cases and occurs because of failure of optic vesicle development at approximately 22 to 27 days of gestation. Secondary anophthalmos is lethal and occurs when the entire anterior neural tube fails to develop. Degenerative or consecutive anophthalmos occurs when the optic vesicles form but subsequently degenerate; consequently neuroectodermal elements may be present in degenerative anophthalmos but are absent in primary and secondary anophthalmos.
Microphthalmos refers to a small ocular globe with an ocular total axial length (TAL) 2 standard deviations less than that of the population age-adjusted mean. Microphthalmos is further classified as severe (TAL <10 mm at birth or <12 mm after 1 year of age), simple, or complex depending on the anatomic appearance of the globe and the degree of TAL reduction. Simple microphthalmos refers to an intact globe with mildly decreased TAL. Complex microphthalmos refers to a globe with anterior segment dysgenesis (developmental abnormalities of the globe anterior to the lens) and/or posterior segment dysgenesis (developmental abnormalities of the globe posterior to the lens) with mild, moderate, or severe decrease in TAL. Severe microphthalmos may be difficult to differentiate from anophthalmia. Both severe microphthalmos and degenerative anophthalmos contain neuroectodermal tissues and are considered as entities along a continuum. The diagnosis is usually based on clinical and imaging criteria.
US is used to determine the ocular TAL in microphthalmos. CT and MR imaging demonstrate an absent globe in anophthalmos ( Fig. 2 ). Amorphous tissue with intermediate density on CT, intermediate signal on T1-weighted images, and low signal on T2-weighted MR images may be noted particularly in degenerative anophthalmos. Orbital dimensions and volumes are reduced in anophthalmos and usually in microphthalmos, unless associated with an intraorbital cyst ( Figs. 3 and 4 ). Simple microphthalmos demonstrates a normal albeit small globe, with normal signal characteristics.
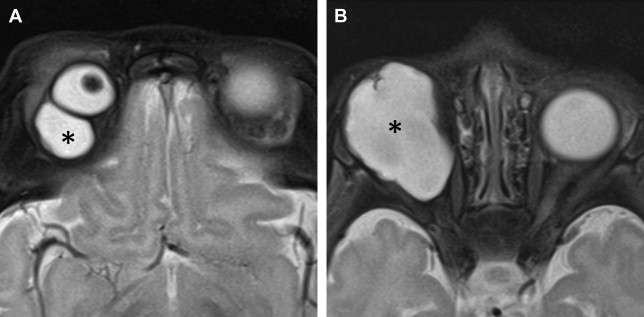
Mild to moderate microphthalmia is managed conservatively with conformers, whereas severe microphthalmia and anophthalmia are treated with endo-orbital volume replacements (implants, expanders, and dermis-fat grafts) and soft-tissue reconstruction.
Cryptophthalmos
Embryologically, the eyelid folds appear during the seventh week and grow toward each other and fuse, with separation of lids occurring between the fifth and seventh months of development. Cryptophthalmos is usually syndromic and results from failure of development of the eyelid folds with absence of eyebrows, eyelids, and the cornea and continuous skin extending from the forehead to the cheeks. Imaging is required to demonstrate the status of the underlying ocular globes and orbital structures before surgical intervention (see Fig. 3 ).
Anterior Segment Dysgenesis
Anterior segment dysgenesis results from faulty development of the anterior ocular structures, including the cornea, iris, ciliary body, lens, and anterior and posterior chambers, resulting in an increased risk of glaucoma and blindness. Anterior segment anomalies are associated with PITX2, FOXC1 PAX6 and related mutations and produce findings such as aniridia, iris hypoplasia, primary congenital glaucoma, Axenfeld-Rieger syndrome (congenital angle anomalies with iris strands), and Peter anomaly (corneal clouding with adhesions between iris, lens, and cornea). Although many of these anomalies produce ophthalmologic abnormalities that do not require further imaging, thin-section high-resolution T2-weighted 3T MR images may demonstrate abnormal size and shape of the anterior and posterior chambers and/or buphthalmos ( Fig. 5 ).
Macrophthalmos
Elongation of the anteroposterior (AP) diameter of the posterior chamber of the globe is most frequently caused by severe myopia. Buphthalmos refers to diffuse enlargement of the AP diameter of the globe (anterior and posterior chambers) usually due to primary congenital or infantile glaucoma or due to syndromic glaucoma as seen in neurofibromatosis type I (NF-1) and Sturge-Weber syndrome.
Imaging is required to differentiate macrophthalmos from other conditions resulting in enlargement of the globe such as an intraocular mass. In buphthalmos, the globe is generally uniformly enlarged but may occasionally have oval or bizarre configurations. Patients with NF-1 demonstrate proptosis, sphenoid wing dysplasia, and orbital plexiform neurofibromas in addition to buphthalmos ( Fig. 6 ).
Congenital Cystic Eye
Congenital cystic eye is a rare condition resulting from failure of the optic vesicle to invaginate to form the globe. On imaging, a cystic, sometimes septated, orbital mass is seen in place of the normal globe ( Fig. 7 ). The differential diagnosis includes microphthalmia with cyst, microphthalmia with cystic teratoma, ectopic brain tissue, and meningoencephalocele.
Coloboma
Coloboma is a developmental anomaly that results from incomplete closure of the embryonic choroidal fissure, resulting in ectasia and herniation of vitreous into the retro-ocular space. The developmental insult occurs during gestational days 35 to 41. Colobomas may affect the iris, lens, ciliary body, retina, choroid, sclera, or optic nerve. Colobomas may be unilateral or bilateral and can be isolated or syndromic, as seen in CHARGE syndrome (coloboma, heart defects, choanal atresia, retarded growth and development, genital malformations, and ear anomalies). Numerous other genetic disorders are associated with coloboma, including focal dermal hypoplasia, branchio-oculofacial syndrome, trisomies 13 and 18, and Aicardi syndrome.
On MR imaging, coloboma appears as a focal defect of the posterior wall of the globe, sometimes associated with microphthalmia ( Fig. 8 ). A minimal defect results in a small excavation along the posterior globe. A larger defect produces a retrobulbar cystic cavity outpouching from the posterior wall of the globe.
Morning Glory Disc Anomaly
Morning glory disc anomaly (MGDA) is a congenital optic nerve anomaly characterized by a funnel-shaped excavation of the optic disc with a central glial tuft overlying the optic disc and an annulus of chorioretinal pigmentary changes surrounding the optic disc excavation.
Although MGDA is usually diagnosed clinically, imaging also distinguishes MGDA from optic nerve coloboma. MGDA has 3 distinctive MR imaging findings: (1) funnel-shaped appearance of the optic disc with elevation of the adjacent retinal surface; (2) abnormal tissue associated with the ipsilateral distal intraorbital optic nerve, effacing the adjacent perioptic nerve subarachnoid space; and (3) lack of the usual enhancement at the lamina cribrosa associated with the funnel-shaped defect at the optic papilla ( Fig. 9 ). Identification of MGDA at imaging should prompt a search for associated intracranial abnormalities, including midline craniofacial and skull base defects, vascular abnormalities, and cerebral malformations. In particular, brain MR imaging and internal carotid artery MRA should be performed because of an association with transphenoidal basal encephalocele and congenital steno-occlusive change of the internal carotid arteries (moyamoya disease) in these patients. MGDA can also be seen in association with PHACES ( p osterior fossa anomalies, h emangioma, a rterial and aortic arch anomalies, c ardiac anomalies, e ye anomalies, and s ternal anomalies and/or supraumbilical raphe).
Staphyloma
Staphyloma results from thinning and stretching of the uvea and sclera and involves all layers of the globe. Risk factors include severe axial myopia, glaucoma, and severe ocular inflammation. Imaging reveals a posterior outpouching of the globe producing deformity in globe contour ( Fig. 10 ).
Hypertelorism, Hypotelorism, and Cyclopia
Hypertelorism denotes increased distance between the medial orbital walls. Hypertelorism is associated with several craniofacial disorders, including cephaloceles, syndromic agenesis of the corpus callosum, and syndromic coronal craniosynostosis ( Fig. 11 A). Hypertelorism must be distinguished from dystopia canthorum in which the medial orbital walls are normally spaced but the medial intercanthal distance is increased, as seen in various types of Waardenburg syndrome.
Hypotelorism denotes decreased distance between the medial orbital walls. Hypotelorism is also associated with a variety of disorders, including the holoprosencephalies (HPE) and premature fusion of the metopic and sagittal sutures (see Fig. 11 B). Cyclopia or cyclophthalmia denotes complete fusion of the optic vesicles resulting in a single median eye. Synophthalmia represents partial fusion of the optic vesicles resulting in duplication of some anterior structures. Both cyclophthalmia and synophthalmia can be associated with HPE. Other manifestations of HPE include ethmocephaly (hypotelorism with median proboscis) and cebocephaly (hypotelorism with rudimentary nose and single nostril).
Large/Small Orbit
A large orbit can be congenital or acquired and results from causes such as cephalocele, bony deformity as seen in NF-1, and bony or orbital masses (see Figs. 6 and 7 ). A small orbit accompanies anopthalmia and microphthalmia (see Figs. 2 and 3 ). Exorbitism denotes shallow orbits as seen in syndromic craniosynostosis (see Fig. 11 ). Exorbitism should not be confused with proptosis in which there is mass effect within the orbit causing ventral protrusion of the globe.
Optic Nerve Hypoplasia
Optic nerve hypoplasia (ONH) is a developmental anomaly characterized by optic nerve underdevelopment. Bilateral ONH is associated with syndromic disorders such as septo-optic dysplasia. On imaging, the affected optic nerves and part or all of the optic chiasm and tracts appear small (see Fig. 10 ). The differential diagnosis for optic hypoplasia is optic atrophy resulting from a variety of causes such as prior infection/inflammation, trauma, irradiation, retinopathy of prematurity (ROP), and vascular insult.
Persistent Hyperplastic Primary Vitreous
Persistent hyperplastic primary vitreous (PHPV) results from failure of the embryonic hyaloid vasculature to involute, resulting in persistence of hyperplastic primary vitreous and the capillary vascular network covering parts of the lens. PHPV is typically unilateral and results in congenital microphthalmos, leukocoria, and cataract. Bilateral PHPV is associated with congenital conditions such as Norrie disease and Warburg disease.
The appearance of PHPV has been likened to that of a martini glass with the glass represented by triangular retrolental fibrovascular tissue and the martini glass stem represented by the stalk of hyaloid remnant extending to the optic disc in Cloquet canal ( Fig. 12 ). On CT, PHPV appears as increased density of the vitreous with a V-shaped or linear structure presumed to represent a remnant of the Cloquet canal. On MR imaging, the retrolental fibrovascular tissue and stalklike hyaloid remnant are hypointense on T1- and T2-weighted images with contrast enhancement. Hemorrhage and layering vitreous debris may be seen on imaging. Absence of calcification on CT is an important distinction from retinoblastoma ( Box 1 ).
Normal-sized eye
Calcified mass
- •
Retinoblastoma (single or multiple enhancing lesions, grows into vitreous or choroid)
- •
Noncalcified mass
- •
Coats disease (no enhancing mass, lipoproteinaceous hyperintense subretinal exudate)
- •
Microphthalmia
Unilateral
- •
PHPV (subhyaloid or subretinal blood-fluid levels, retrolental tubular mass along the hyaloid canal that enhances)
- •
Bilateral
- •
ROP (no or minimal enhancement, dystrophic calcification late in disease)
- •
Bilateral PHPV (see earlier)
- •
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


