Clinical Presentation
The patient is a 16-year-old male who presented with history of a “cold and sore throat that lasted a month” followed by progressive acute lower extremity weakness. This was followed by numbness in the toes and fingers and later dizziness. On physical examination, he had absent lower extremity reflexes and reduced strength in the feet, toes, hip flexors, deltoid, and finger flexors. He was observed to be ataxic and had a mild upper extremity tremor. A magnetic resonance (MR) scan of the lumbar spine was ordered. Nerve conduction studies revealed findings consistent with a demyelinating polyradiculomyelopathy. The protein level in the cerebrospinal fluid (CSF) was elevated (115 mg/dL). A diagnosis of Guillain-Barré syndrome was made. He was treated with intravenous immunoglobulin (IVIg), and symptoms improved but did not resolve completely after several months of follow-up.
Imaging Presentation
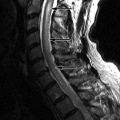
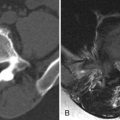
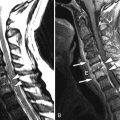
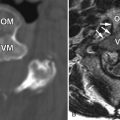
MR imaging of the lumbar spine obtained at time of presentation revealed diffuse thickening and enhancement of the nerve rootlets of the cauda equina and nerve roots consistent with diffuse inflammatory polyneuritis in keeping with the clinical diagnosis ( Figs. 38-1 to 38-4 ) .



Discussion
Diffuse inflammatory polyneuropathies of the cauda equina, spinal nerve roots, and peripheral nerves represent a diverse group of disorders with many similarities in terms of pathogenesis, clinical presentation, biochemical properties, and histologic origins and in terms of similarities seen on imaging studies. Acute polyneuropathy or polyradiculoneuropathy is usually classified under the general category of Guillain-Barré syndrome (GBS) . GBS is the most common cause of acute muscle weakness in patients under age 40, and its incidence is 1 to 2 per 100,000 people. GBS most commonly occurs in children and young adults. Chronic inflammatory polyradiculoneuropathy (CIPD) is a condition that usually has a different presentation and clinical course. However, GBS may progress to CIDP or resemble CIDP clinically in the later stages of the disease.
There are at least six clinical subtypes of Guillain-Barré syndrome : (1) Acute inflammatory demyelinating polyradiculoneuropathy ( AIDP ) is often used synonymously with GBS because AIDP is by far the most common subtype of GBS. AIDP is a post-infectious or post-vaccination, immune-mediated process that attacks the Schwann cells, causing neural inflammation and demyelination that may involve the cauda equina, nerve roots, peripheral nerves, and/or cranial nerves. AIDP classically manifests as an ascending paralysis beginning in the distal lower extremities and extending cephalad to a varying degree in thethorax, cervical region, and sometimes into the brainstem. (2) Acute motor axonal neuropathy ( AMAN ) , also called Chinese paralytic syndrome , attacks motor nerves at the nodes of Ranvier, which are gaps in the myelin sheath between adjacent Schwann cells. AMAN is prevalent in China and Mexico. (3) Acute motor sensory axonal neuropathy ( AMSAN ) is similar to AMAN with motor dysfunction, but also causes severe axonal dysfunction of sensory nerves. (4) Miller-Fisher syndrome ( MFS ) is a rare form of GBS with patients presenting initially with opthalmoplegia, ataxia, and areflexia. A descending pattern of paralysis occurs in MFS. (5) Bickerstaff’s brainstem encephalitis ( BBE ) is believed to be a variant of GBS characterized by acute onset of opthalmoplegia, ataxia, disturbance of consciousness, hyperreflexia, and a positive Babinski sign. (6) Acute panautonomic neuropathy ( APAN ) is the rarest form of GBS, which manifests as diffuse autonomic nervous system dysfunction. Patients present with sympathetic and parasympathetic nervous system dysfunction, cardiac dysrhythmias, orthostatic hypotension, and sometimes encephalopathy. APAN has a high mortality rate.
GBS is an acute motor and sensory axonal neuropathy. Symptoms usually develop after a viral illness or after vaccination. Patients present with lower extremity flaccid paralysis or distal lower extremity paresthesias (numbness and tingling), hyporeflexia, or areflexia. This is followed by ascending paralysis . Eventually the paralysis may involve the brainstem, which causes respiratory paralysis requiring ventilator support. Autonomic nerve dysfunction is common. Cranial nerve palsies may occur in up to one half of patients, facial nerve paralysis being the most common.
Neuropathic pain is not considered a common primary feature of GBS but does develop in many patients, and the pain can be severe. However, many types of pain can occur in patients with GBS. In one series of 55 GBS patients, nearly one half of the patients presented with moderate to severe pain. The back and leg pain usually resolves by 8 weeks after onset, but dysesthetic pain in the extremities may persist. GBS must always be considered in the differential diagnosis of children presenting with back or leg pain. The extremity pain associated with GBS is often worse at night and can be a deep muscular aching pain or spasmodic pain, described commonly as a charley horse . Painful dysesthesia may also occur similar to that commonly seen with diabetic neuropathy. Patients with GBS may also experience abdominal pain. Muscle weakness and fatigue may be prominent features in more chronic cases of GBS. Patients with GBS typically have elevated protein in the CSF. The majority of patients with GBS show improvement in 2 to 3 months, but up to 50% have persistent symptoms at 1 year follow-up. Permanent deficits remain in up to 10% of cases. Up to 8% of AIDP cases can be fatal. GBS may evolve into a condition that is clinically similar to or indistinguishable from CIDP. The diagnosis of CIDP should be considered if a patient with GBS deteriorates after 9 weeks from onset of symptoms or when the patient’s condition deteriorates three times or more.
GBS is believed to be an immune-mediated process that causes inflammatory demyelinating lesions in the cauda equina, nerve roots, peripheral nerves, or cranial nerves. Antibodies to ganglioside GM1 (anti-GM1 antibodies) are elevated in the serum and CSF of patients with GBS and AMAN. Anti-GM1 antibodies have also been implicated in the pathogenesis of amyotrophic lateral sclerosis (ALS). The GM1 epitope is present in motor neurons and their axons, in the dorsal root ganglia, and in sensory axons. The precise role of anti-GM1 antibodies in the pathogenesis of GBS is still not fully known. There is some evidence that patients with GBS who have anti-GM1 antibodies more often experience a rapidly progressive and more severe neuropathy with predominantly distal distribution of weakness. Other autoantibodies may also be involved in the pathogenesis of GBS. For example, patients with acute polyneuropathies may have elevated titers of IgG autoantibodies against disialosyl epitopes. Histologically, patients have focal segmental demyelination, perivascular and endoneural lymphocytic infiltrates, and macrophage infiltrates; axonal degeneration may eventually occur.
A Campylobacter jejuni infection can trigger GBS. Patients with C. jejuni infection more commonly experience severe pure motor deficits. In one study, patients with C. jejuni infections responded better to intravenous immunoglobulin (IVIg) therapy than to plasma exchange.
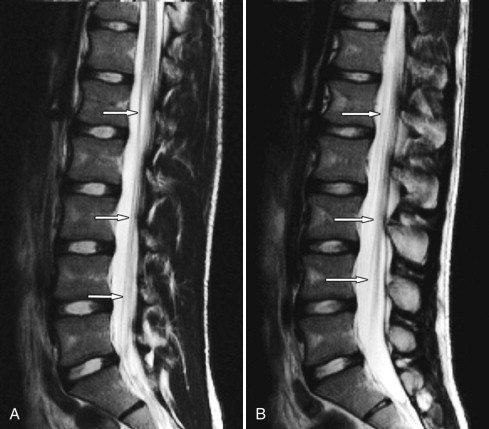
Chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) is an acquired immune-mediated demyelinating disorder involving spinal nerve roots, cauda equina ( Figs. 38-5 to 38-10 ) , lumbar plexus, brachial plexus, or peripheral nerve roots. The prevalence of CIDP is probably grossly underestimated. The pathogenesis of CIDP is unknown but is believed to be related to both abnormal humoral and cellular immune responses to some nerve antigen or antigens. Activated T cells are involved in the generation of inflammatory lesions in the nervous system in general and are also involved in CIDP. The CD4 and CD8 T-cell subgroups have both been implicated in this process. The same type of invariant T-cell found in patients with multiple sclerosis is also present in the majority of patients with CIDP. Regardless of the precise immunologic mechanism, T-lymphocytes, monocytes, and macrophages infiltrate the nerves and adjacent tissue and cause release of enzymes that cause inflammation in the nerves. Soluble cytokines and chemokines also contribute to the pathogenesis of CIDP, and concentrations of these substances are elevated in the CSF in patients with CIDP.






Histologically, the nerves in patients with CIDP show segmental demyelination and remyelination and thinning of myelin sheaths, and the nerves often have an onion bulb appearance, features present in many of the chronic polyneuropathies. Mononuclear cell infiltrates may be present in the endoneurium. There may be a preferential involvement of the motor nerves, which may account for negative sural nerve biopsies in some patients.
Most cases of CIDP are idiopathic, but it can be associated with a variety of disorders including diabetes, HIV infection, celiac disease (gluten-sensitive enteropathy), melanoma, Sjögren’s syndrome, lymphoma, IgM gammopathy (polyclonal or monoclonal), or with hepatitis. Patients with chronic polyneuropathies often have elevated antiganglioside and/or antisulfatide IgM antibodies. The peripheral nerves are most commonly involved in patients with CIDP. Biopsy of the sural nerve may help confirm the diagnosis. In the spine, CIDP affects the cauda equina and lumbar nerve roots more commonly than the cervical nerve roots. The thoracic nerve roots and cranial nerves may also be affected but are less commonly involved than in AIDP (see Fig. 38-7 ).
Patients with CIDP classically present with a history of a chronic, progressive, or relapsing disorder with muscle weakness and often sensory loss. Patients with sensory neuropathy have predominant involvement of large-fiber nerves. This is usually a polyradicular condition with symmetric or asymmetric involvement of proximal and distal muscles in the lower extremity. Neurogenic pain is not a common initial manifestation of CIDP, but pain may occur as the disease evolves. Rarely, neurogenic pain is the initial manifestation of CIDP. Neuropathic pain is very common in patients with small-fiber peripheral neuropathies, such as patients with diabetic neuropathy or amyloidosis-related neuropathy. Patients may present with symptoms similar to multiple sclerosis.
Classically, GBS symptoms progress within the first few weeks and are maximal at 4 weeks from onset of the disease, and thereafter the disorder is usually monophasic. In CIDP, symptoms usually progress for at least 2 months, and thereafter the disorder may be monophasic, relapsing-remitting, or steadily progressive. In practice, however, it may be difficult clinically to differentiate CIDP from progressive GBS, especially in the early stages of the disease. The major electromyographic (EMG) finding in CIDP is slow nerve conduction similar to that seen with demyelinating disorders in general. A more detailed summary of the EMG findings of CIDP can be found elsewhere. Patients with mild cases of CIDP tend to recover with complete or near complete regaining of function. Severe cases have a chronic, progressive, relapsing-remitting illness. Rarely, CIDP can be fatal.
Sensory ganglioneuropathies , which involve the dorsal root ganglia, represent a subgroup of polyneuropathies that may occur with paraneoplastic subacute sensory neuronopathy (SSN), Sjögren’s syndrome, Miller-Fisher syndrome, Bickerstaff’s brainstem encephalitis, inherited ganglioneuropathies (Friedreich’s ataxia and other inherited cerebellar ataxias), and with certain drugs (pyridoxine, vincristine, taxane, and cisplatin). There is a predisposition to developing chemotherapy-induced neuropathy in patients who are diabetic, chronic alcoholics, and in patients with inherited neuropathies.
Paraneoplastic neuropathy (PPN) is rarely encountered in routine practice but may produce clinical manifestations and an MR imaging appearance identical to GBS or CIDP. Indeed, PPN may be considered to be a subtype of CIDP. There are other paraneoplastic syndromes that affect the nervous system, including paraneoplastic encephalomyelitis, paraneoplastic cerebellar degeneration, cancer-associated retinopathy, and paraneoplastic opsoclonus-myoclonus-ataxia syndrome. Paraneoplastic neuropathy commonly involves the dorsal root ganglia, but the spinal cauda equina, nerve roots, and peripheral nerves may be involved as well ( Fig. 38-11 ) .