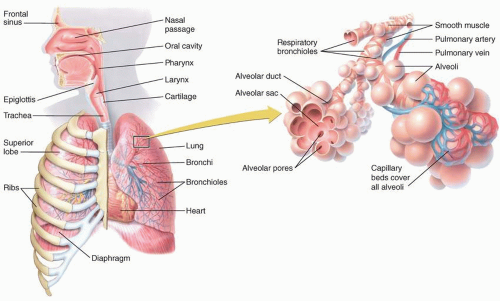
 FIGURE 2.1. Diagram of the large airways, which can be classified as either upper or lower airways. (Adapted from McArdle WD, Katch FI, Katch VL. Exercise Physiology; 2014. Figure 12.1. © LWW/Wolters Kluwer, with permission.) |
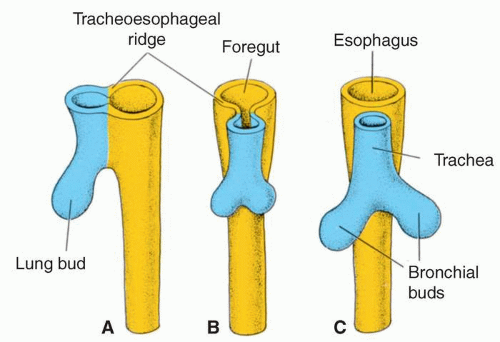 FIGURE 2.2. Diagram of development of the lower airways. The lower airways develop in successive stages from the primitive foregut: A: First, a respiratory diverticulum or “lung bud” is formed ventrally. B: Subsequently, the respiratory diverticulum is separated from the foregut (dorsal) by the tracheoesophageal ridges that join to form a septum. C: Finally, the respiratory diverticulum develops into the trachea and bronchial buds and the foregut forms the esophagus. (Adapted from Sadler TJ. Langman’s Essential Medical Embryology; 2004. Figure 6.1. © Wolters Kluwer, with permission.) |
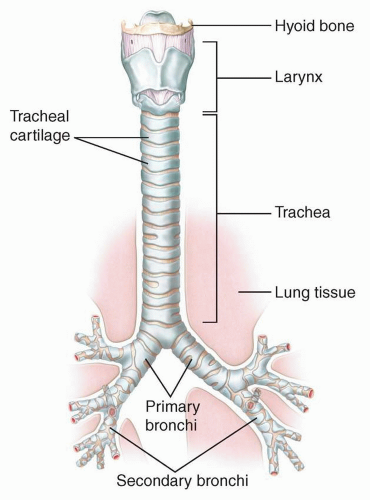 FIGURE 2.3. Anatomy of the trachea. (Modified from Premkumar K. Anatomy & Physiology: The Massage Connection. Baltimore, MD: LWW/Wolters Kluwer; 2004, with permission.) |
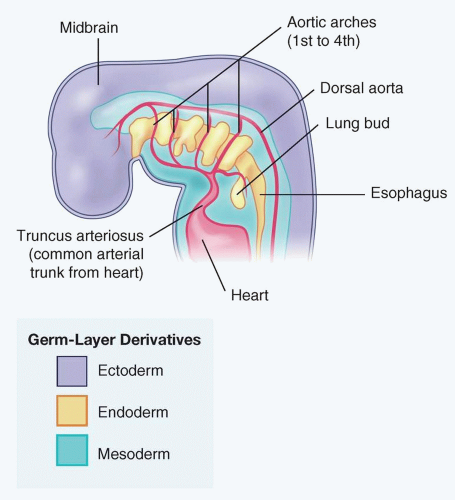 FIGURE 2.4. Developmental embryology of lung buds. |
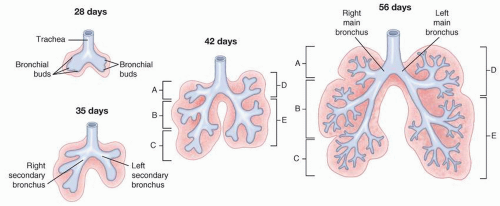 FIGURE 2.5. Development of the distal airways. A: Right upper (superior) lobe. B: Right middle lobe. C: Right lower (inferior) lobe. D: Left upper (superior) lobe. E: Left lower (inferior) lobe. |
standing position.3 Evaluation for air trapping, a secondary sign of tracheobronchial foreign body obstruction, can be further evaluated with expiratory views in cooperative older children or the less technically onerous lateral decubitus views in younger uncooperative patients.3 Gonadal shielding can help limit radiation exposure.
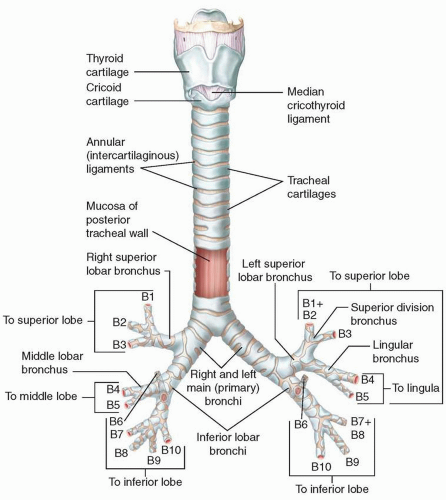 FIGURE 2.6. Trachea and major bronchi of the lungs. |
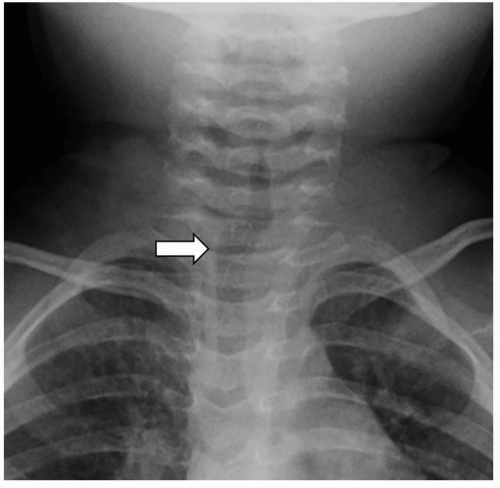 FIGURE 2.7. A 2-year-old girl with normal lateral deviation of the trachea. Frontal radiograph shows normal deviation (arrow) of the trachea to the right of midline at the thoracic inlet level. |
airway from nasopharynx to bronchi can be evaluated rapidly and noninvasively in any projection. Radiation-dose reduction techniques such as pulsed fluoroscopy and restricted fluoroscopic time should be emphasized to keep exposure “As Low As Reasonably Achievable” (ALARA). Uncooperative infants and young children and older children with large body habitus may prove difficult to image.3,7
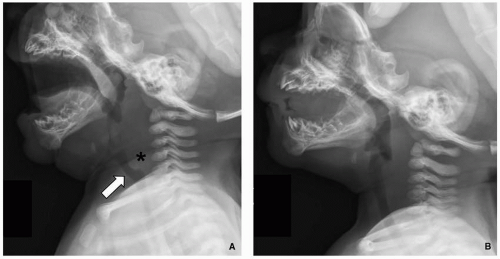 FIGURE 2.8. A 2-year-old boy with normal anterior buckling of the trachea. A: Lateral soft tissue neck radiograph shows anterior buckling (arrow) of the trachea and retropharyngeal soft tissue widening (asterisk) during expiration and neck flexion. B: Repeat lateral soft tissue neck radiograph demonstrates resolution of anterior buckling of the trachea and retropharyngeal soft tissue widening during inspiration. |
TABLE 2.1 Radiograph Technique for Soft Tissue Neck | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
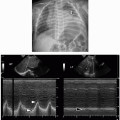
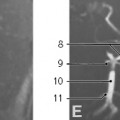
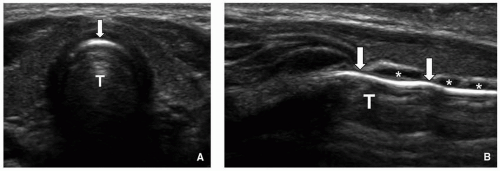 FIGURE 2.9. A 3-year-old boy with normal sonographic view of the trachea. A: Transverse ultrasound image of the trachea obtained in supine position shows air within the nondependent portion of the trachea (T), manifested as a curved hyperechoic line (arrow) with posterior shadowing. B: Sagittal ultrasound image of the trachea obtained in supine position demonstrates air as a hyperechoic line (arrows) within the nondependent portion of the trachea (T). Asterisks demarcate tracheal cartilages. |
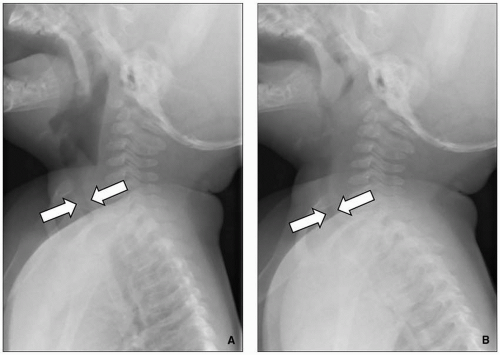 FIGURE 2.10. A 6-month-old girl with bronchoscopy-confirmed tracheomalacia who presented with chronic cough. A: Lateral radiograph obtained at end-inspiration during airway fluoroscopy shows a patent trachea (arrows). B: Lateral radiograph obtained at end-expiration during airway fluoroscopy demonstrates substantial (>50%) collapse of the trachea (arrows), consistent with tracheomalacia. |
milliamperage (mA), lowest-possible kilovoltage (kV), and anatomically based real-time automated exposure control help to decrease radiation exposure (Table 2.2). The intrinsic contrast between the air-filled large airways and nearby mediastinal soft tissue structures also allows for lower-dose technique. In addition to traditional coronal and sagittal reconstructions, curved planar reformats along the long axis of the large airways are commonly performed to allow more accurate measurements of the trachea and bronchi.3,7,12,16
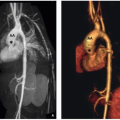
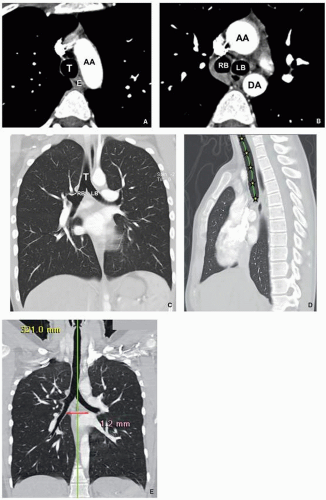 FIGURE 2.11. A 10-year-old boy with normal large airways. A: Axial contrast-enhanced CT image at the level of the aortic arch (AA) shows a normal, round, patent trachea (T) at end-inspiration. E, Esophagus. B: Axial contrast-enhanced CT image at the level of the proximal mainstem bronchi demonstrates a normal, patent right mainstem bronchus (RB) and left mainstem bronchus (LB). AA, Ascending aorta; DA, Descending aorta. C: Coronal lung window CT image shows the trachea (T) and bilateral mainstem bronchi. RB, Right mainstem bronchus; LB, Left mainstem bronchus. D: Sagittal reformatted lung window CT image demonstrates a reference line (green line and yellow stars) through the center of the airway for reconstruction of a curved coronal reformatted CT image. E: Curved coronal reformatted CT image shows a straightened view of the large airways. |
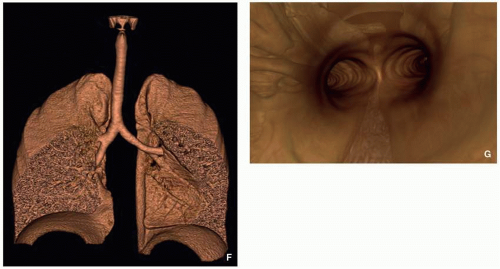 FIGURE 2.11. (Continued) F: Three-dimensional volume-rendered CT image (virtual bronchography) shows the normal large airways. G: Three-dimensional volume-rendered CT image (virtual bronchoscopy) of the normal large airways at the level of carina shows patent bilateral mainstem bronchi. |
albumin, and ferumoxytol (Feraheme), an iron oxide nanoparticle, have shown excellent promise as vascular contrast agents, although use remains off-label, and gadofosveset is no longer being marketed. Moreover, recently, heightened safety concern has been raised about ferumoxytol, which is approved only for the treatment of iron deficiency anemia. Nevertheless, ferumoxytol is not excreted through the kidneys and remains an attractive contrast option for patients with renal failure.18
TABLE 2.2 Tube Current and kV by Patient Weight for Central Airway MDCT | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||
TABLE 2.3 Static MRI Airway Imaging Protocol | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||
TABLE 2.4 Dynamic MRI Airway Imaging Protocol | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||
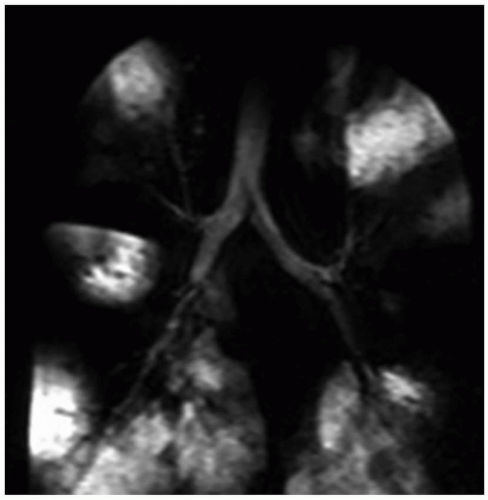 FIGURE 2.12. Hyperpolarized 3He MR lung ventilation image in a patient with cystic fibrosis causing severe respiratory disease (FEV1 = 34%). In addition to wedge-shaped regions of hypointensity due to underlying lung disease, large airways are also visualized. (Reprinted from Liszewski MC, Hersman FW, Altes TA, et al. Magnetic resonance imaging of pediatric lung parenchyma, airways, vasculature, ventilation, and perfusion: state of the art. Radiol Clin North Am. 2013;51(4):573, with permission.) |
tongue. The patient is dynamically imaged using the gamma camera. Aspiration is suggested by the presence of radiotracer within the tracheobronchial tree. Relatively sensitive and safe, the radionuclide salivagram can be performed in children who are not yet feeding orally, and it requires minimal patient cooperation.26 It is thus a viable alternative to the fluoroscopic modified barium swallow study. As in other body regions, positron emission tomography (PET) using 18F-fluorodeoxyglucose (18F-FDG), combined with anatomical imaging, provides added diagnostic value in characterizing neoplastic, inflammatory, and infectious processes of the airways.27
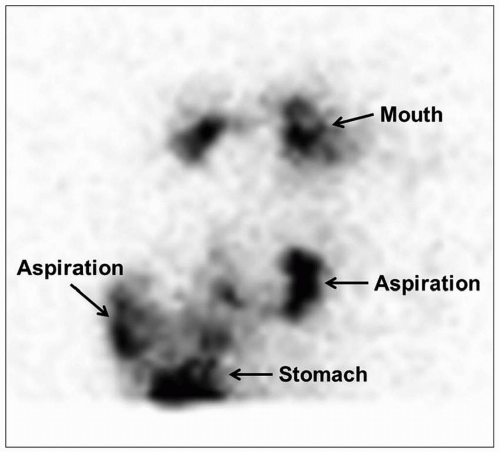 FIGURE 2.13. A 2-year-old girl with developmental delay and aspiration. Salivagram shows aspiration into both mainstem bronchi. |
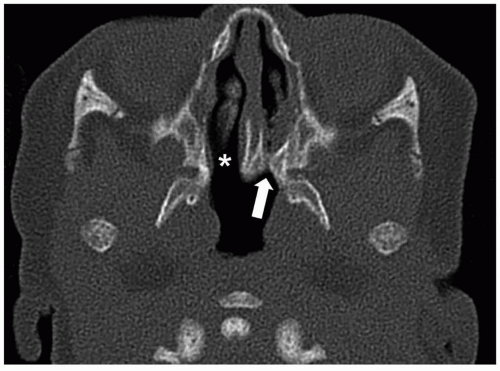 FIGURE 2.14. A 2-day-old boy with choanal atresia who presented with grunting and clinical failure to pass a nasogastric tube through the left nostril. Axial bone window CT image at the level of the choanae shows complete obliteration of the left nasal passage (arrow) compared to the open contralateral side (asterisk). |
inside the nares, overgrowth and medial displacement of the nasal processes of maxilla, and narrowing of the pyriform aperture (Fig. 2.15). The pyriform aperture is bounded by the nasal bones superiorly, the nasal processes of the maxilla laterally, and the horizontal processes inferiorly; a width <11 mm in a term infant is considered diagnostic of CNPAS.7,36,38,39 MR is used to assess for associated intracranial abnormalities.40
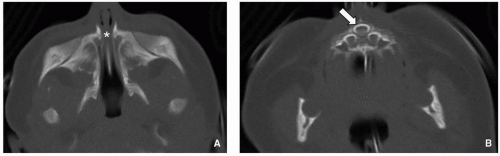 FIGURE 2.15. A 1-day-old girl with congenital pyriform aperture stenosis and midline incisor who presented with grunting, difficulty breathing, and clinical failure to pass a nasogastric tube on either side. An orogastric tube was inserted instead. A: Axial bone window CT image obtained at the level of the nostrils shows marked narrowing (asterisk) of the pyriform aperture. B: Axial bone window CT image obtained at the level of the maxillary alveolar ridge demonstrates a solitary median maxillary central incisor (arrow), a commonly associated finding in patients with congenital pyriform aperture stenosis. |
 FIGURE 2.16. A 12-year-old girl with tonsillar hyperplasia. The cut surfaces of the resected palatine tonsils show fleshy pale tan lymphoid tissue with deep slit-like crypts. |
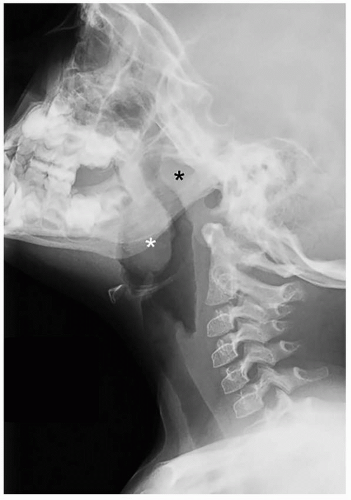 FIGURE 2.17. A 12-year-old boy with adenoid and palatine tonsillar hypertrophy who presented with sleep apnea. Lateral soft tissue neck radiograph shows moderate adenoid (black asterisk) and palatine tonsillar (white asterisk) hypertrophy. |
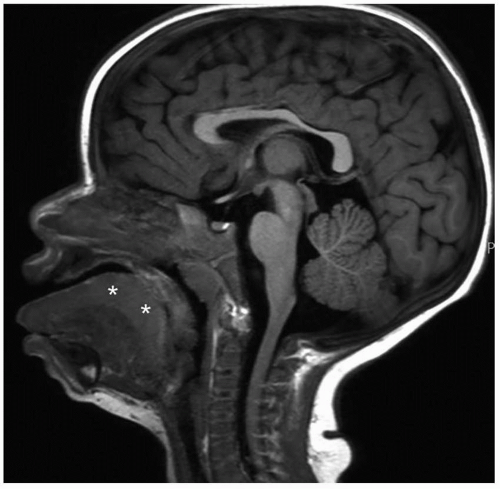 FIGURE 2.18. A 3-year-old girl with macroglossia who presented with dysphagia and drooling. Sagittal T1-weighted magnetic resonance image shows marked enlargement of the tongue (asterisks) with anterior protrusion outside of the oral cavity. |
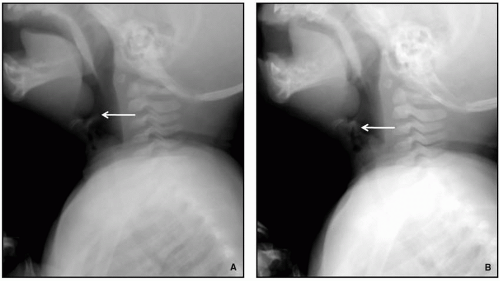 FIGURE 2.19. A 2-month-old girl with laryngomalacia who presented with stridor. A: Lateral soft tissue neck radiograph shows the normal position of the epiglottis (arrow). B: Lateral soft tissue neck radiograph demonstrates laxity of the epiglottis (arrow), with posterior and downward movement obstructing the airway. (Reprinted from Laya BF, Lee EY. Congenital causes of upper airway obstruction in pediatric patients: updated imaging techniques and review of imaging findings. Semin Roentgenol. 2012;47[2]:147-158, with permission. Case courtesy of Khristine Grace C. Pulido, MD, Manila, Philippines.) |
the lower lobes (Fig. 2.21).63,64 Chest CT demonstrates similar findings, but with greater sensitivity and anatomic precision (Fig. 2.22). Modified barium swallow study is useful for assessing aspiration. Barium swallow study may show passage of contrast into the trachea, potentially leading to misdiagnosis of tracheoesophageal fistula (TEF) rather than laryngeal cleft.63,64 CT or MRI may occasionally demonstrate the abnormal communication and lack of soft tissue between the trachea and esophagus. An abnormally anterior or intratracheal position of a nasogastric tube is also a clue to the diagnosis. Ultimately, advanced modalities are more often used for assessing associated anomalies.63,64
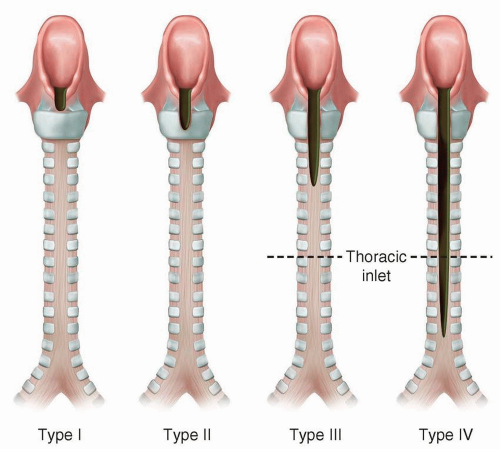 FIGURE 2.20. Diagram showing the types of laryngeal cleft. Type I is the mildest form of laryngeal cleft. The gap between the larynx and the esophagus is located above the vocal cords. Type II laryngeal cleft extends into the lower cartilage of the larynx, below the vocal chords. Type III laryngeal cleft extends beyond the larynx and into the trachea. Type IV is the most severe form of laryngeal cleft. The gap extends even further down into the trachea, even sometimes extending to the carina. |
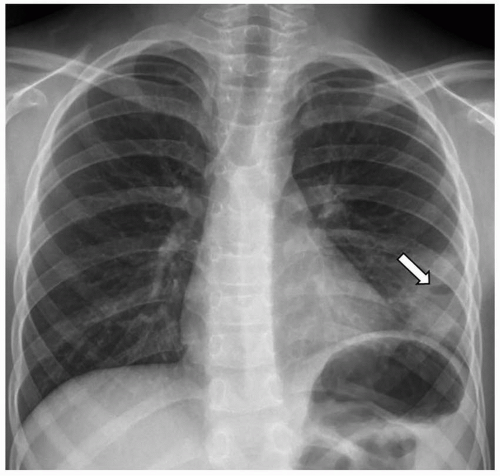 FIGURE 2.21. A 10-year-old boy with delayed diagnosis of type II laryngeal cleft who presented with recurrent pneumonia. Frontal chest radiograph shows a consolidation and an internal air-fluid level (arrow), likely representing a lung abscess caused by aspiration. |
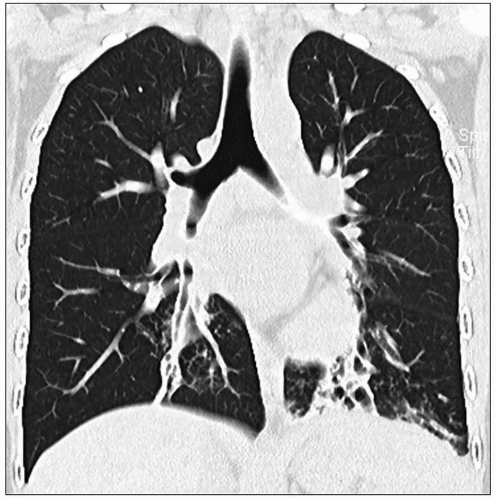 FIGURE 2.22. A 7-year-old boy with delayed diagnosis of type II laryngeal cleft who presented with recurrent aspiration pneumonia. Coronal lung window CT image shows bronchiectasis and chronicappearing atelectasis in both lower lobes, left side greater than right side, due to recurrent aspiration pneumonia. |
choice, as it is able to show the entire length of the atretic trachea and precisely localize the esophageal fistula.65,69
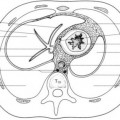
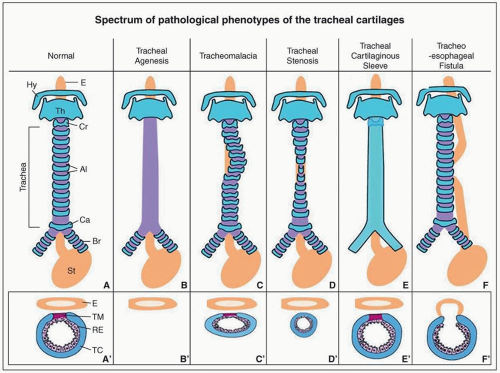 FIGURE 2.23. Spectrum of pathological phenotypes of the tracheal cartilages showing normal anatomy (A-F) and cross-section through the tracheal cartilages (A’-F’). Cartilages are depicted in blue, epithelium in purple, and esophagus/stomach in orange. E, esophagus; Hy, hyoid; Th, thyroid cartilage; Cr, cricoid cartilage; Al, annular ligaments; Ca, carina; Br, bronchus; St, stomach. A’: Schematic cross-section through tracheal cartilages. E, esophagus; TM, trachealis muscle; RE, respiratory epithelium; TC, tracheal cartilage. B and B’: Depiction of tracheal agenesis shows absence of the tracheal cartilages below the larynx. C and C’: Tracheomalacia results from a weakness in the cartilage rings that makes the trachea prone to collapse. This can be seen as a flattening of the tracheal rings in cross-section (C’). D and D’: Tracheal stenosis is a narrowing of the tracheal lumen. Rather than the normal C-shaped cartilage rings with intervening tissue allowing expansion, in tracheal stenosis the cartilage rings are O-shaped, and therefore unable to grow. E and E’: In tracheal cartilaginous sleeve, cartilage rings are fused, leading to a loss of the intervening fibrous annular ligaments. F and F’: Tracheo-esophageal fistula occurs when the foregut fails to separate into the trachea and esophagus. (Reprinted from Sher ZA, Liu KJ. Congenital tracheal defects: embryonic development and animal models. AIMS Genetics. 2016;3(1):60-73. Figure 1, with permission.) |
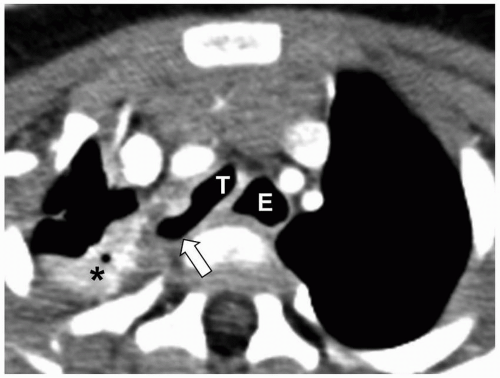 FIGURE 2.24. A 2-year-old girl with tracheal bronchus who presented with recurrent right upper lobe pneumonia. Axial enhanced CT image shows a tracheal bronchus (arrow) arising at the right lateral aspect of the trachea (T) and leading to the consolidated right upper lobe (asterisk). E, Esophagus. |
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


