Abstract
Cystic hygroma (CH) is a relatively common malformation of the lymphatic system. It is important for clinicians to be familiar with the condition because it is one of the few developmental abnormalities that can be reliably diagnosed during the first-trimester sonogram. In addition to intrinsically contributing to fetal/neonatal morbidity and mortality, CH and other lymphatic malformations are associated with a dramatic increase in the risk for aneuploidy, other genetic abnormalities and syndromes, and other birth defects. This chapter will highlight the diagnosis and management of fetuses with CH and other lymphatic abnormalities.
Keywords
cystic hygroma, lymphedema, nuchal translucency, nonimmune hydrops, lymphatic disorders
Background
Dysregulation of the lymphatic system is a key factor in a variety of disorders, and may be associated with aneuploidy (especially monosomy X), autosomal, or X-linked genetic disorders, and/or multiple other conditions. Lymphatic disorders can be classified as primary or secondary. Primary or congenital lymphedema is most commonly associated with cystic hygroma (CH), and often leads to hydrops fetalis and fetal demise. Secondary lymphedema is typically the result of trauma, infection, or radiation treatment for cancer. This chapter will focus on primary (congenital) lymphedema, especially the most common form of lymphatic malformation, CH.
Definition and Associated Conditions
Lymphedema, an accumulation of fluid in interstitial spaces, is characterized by an anatomic or functional defect that causes an imbalance between the rate of lymph production in tissues and removal via the lymphatic system. This results in an array of clinical outcomes ranging from mild cosmetic problems to serious genetic conditions. CH, the most common form of lymphatic abnormality, can lead to nonimmune hydrops and pregnancy loss. It is also discussed in Chapter 71 .
Cystic Hygroma: is caused by a defect in communication between the lymphatic and venous systems leading to localized fluid accumulation within the posterior and posterolateral aspect of the neck, which can eventually progress to fetal hydrops.
Nonimmune Hydrops: are characterized by the presence of fluid in two or more fetal soft tissues and serous cavities. The prevalence ranges from 3 : 10,000–6 : 10,000 births. These include ascites, pleural effusions, pericardial effusions, and generalized skin edema ( Chapter 123 ).
- •
Milroy disease (VEGFR-3): is usually inherited in an autosomal dominant fashion, may also be autosomal recessive and has multiple presentations, including unilateral or bilateral swelling of lower limbs or face with irreversible fibrotic changes, prominent veins, “ski-jump” toenails, and papillomatosis.
- •
Noonan syndrome: can be autosomal dominant inheritance or de novo, with a prevalence of 1 : 10,000–4 : 10,000; is characterized by CH, short limbs, and cardiac defects in addition to lymphedema and pulmonary lymphangiectasia ( Chapter 136 ).
- •
Neurofibromatosis: is an autosomal dominant disorder, with a prevalence of 2.5 : 10,000–3 : 10,000; characterized by a mutation in NF1, presents prenatally with macrocephaly, megalencephaly, café-au lait spots, pseudarthrosis, lisch nodules, and neurofibromas.
- •
Adams-Oliver syndrome: can be transmitted in an autosomal dominant or recessive manner, characterized by congenital cutis aplasia, scalp and skull defects, limb abnormalities, and occasional congenital heart defects.
- •
Hennekam syndrome: is a rare autosomal recessive disorder with only 50 cases reported worldwide; characterized by lymphangiectasia, bilateral pleural effusion, hydrops, facial anomalies, and mental retardation in addition to lymphedema.
- •
Prader-Willi syndrome: is an autosomal recessive disorder that can be caused by a deletion in genes within the paternal arm of chromosome 15, or inheritance of two copies of chromosome 15 of maternal origin, termed uniparental disomy, and rarely translocation. There is a prevalence of 0.3 : 10,000–1 : 10,000 people worldwide. It is characterized by facial anomalies, hypogonadism, small hands and feet, and mental retardation.
- •
Angelman syndrome: is caused by a deletion of maternal arm of chromosome 15 with a prevalence of 0.5 : 10,000–0.8 : 10,000; characterized by microcephaly, neurologic delays with delayed development, severe intellectual disability, and ataxia.
- •
Aagenaes syndrome: an autosomal recessive disorder characterized as childhood growth delay, cholestatic liver disease, and generalized lymphedema.
- •
Lymphedema-distichiasis: is characterized by pubertal onset of peripheral lymphedema and is associated with the presence of a supplemental row of eyelashes (distichiasis) that arise from Meibomian glands.
- •
Hypotrichosis-lymphedema-telangiectasia: has hair and cardiovascular anomalies along with lymphedema (severity associated with hydrops and stillbirth), linked to an abnormal SOX18 gene.
- •
Primary pulmonary lymphangiectasia: is associated with polyhydramnios, bilateral pleural effusions, severe thickening of the small bowel, and ascites. It can occur as a primary disease or as part of other lymphedema syndromes and may be transmitted in an autosomal dominant, recessive, or X-linked manner.
Differential Diagnosis
The differential diagnosis of prenatally detected lymphedema includes teratoma (germ cell tumor with aggressive growth located on the anterior aspect of the neck), occipital encephalocele and meningocele (neural tube defect characterized by protrusions of the membranes of the brain, often associated with craniofacial abnormalities), hemangioma (benign vascular tumor involving the endovascular vessels that usually occurs on face or back, often self-resolving), and goiter ( in utero enlargement of the thyroid).
Diagnosis
A CH, the most common lymphatic abnormality, is a congenital abnormality of the lymphatic system characterized by the presence of fluid-filled spaces (edema) at the sites of lymphatic-venous connection within the posterior neck and back of a fetus. The diagnosis is made by ultrasound (US) findings of an enlarged hypoechoic space at the back of the fetal neck extending along the length of the fetal back. It can also be classified according to the presence or absence of septations; that is, septated versus nonseptated CH. When CH appears septated, the prognosis may be worse than the nonseptated form ( Fig. 121.1 ). However, some studies have shown no difference in outcome between nonseptated and septated forms. Increasing size of the hygroma is associated with relatively worse outcome, regardless of septation. Therefore all fetuses with CHs, regardless of size and septation, require serial evaluation and assessment for associated anomalies.
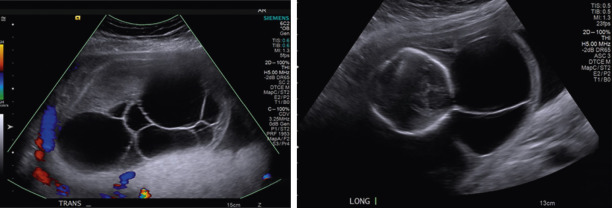
Prevalence
The incidence of prenatally diagnosed CH ranges from 1 : 200 spontaneous abortions, but it is only 1 : 600 to 1 : 700 in low-risk pregnancies later in pregnancy. Data from the First and Second-Trimester Evaluation of Risk Trial showed a prevalence of 1 : 285 in first-trimester pregnancies.
Primary lymphedema is relatively rare, affecting approximately 1 : 10,000 to 1.3 : 10,000 live births. There is a slight female predominance with most of the associated syndromes, with female to male ratios estimated to be between 2.5 : 1 and 10 : 1.
Etiology
Lymphatic malformations are benign vascular lesions that arise from abnormalities in the embryologic development of the lymphatic system. Lymphatic malformations can be seen in any anatomic region but are more common in lymphatic-rich areas such as the head, neck, axilla and mediastinum, abdomen, and retroperitoneum. Cystic lymphatic lesions can be classified as macrocytic, microcytic, or combined. The cause of CH is thought to be a defect in communication between lymphatic vessels, jugular lymph sacs, and jugular veins. This is attributed to an expansion of either irregular embryonic lymphatic tissue or from abnormal growth of lymphatic endothelium between 6 and 9 weeks’ gestation, resulting in deficits within lymphatic vessels. Failure of the jugular lymphatic sacs to attach and drain into the jugular veins leads to lymphatic fluid stasis and progresses to a single or numerous fluid-filled lesions in various locations. This leads to lymph fluid accumulation in the tissue, and in some cases, results in hydrops fetalis.
Primary lymphedema can also manifest as lymphangiectasia of internal organs. As described earlier, pulmonary lymphangiectasia is characterized by pulmonary, subpleural, interlobar, perivascular, and peribronchial lymphatic dilatation, and may be complicated by a chylous pleural effusion. Lymphangiectasia can also occur in the intestines, leading to poor nutritional absorption and severe protein loss with resultant growth restriction.
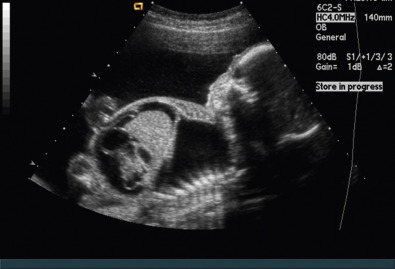
Nonimmune hydrops fetalis is associated with several congenital disorders, including those with lymphatic dysplasia. One of the most common etiologies of hydrops is an imbalance in the regulation of fluid movement between vascular and interstitial spaces, with an increase in interstitial fluid production or a decrease in venous return. The most common etiology of an isolated effusion leading to hydrops is chylothorax ( Fig. 121.2 ) ( Chapter 4 ), which is caused by lymphatic obstruction. The diagnosis is confirmed by the finding of a fetal pleural cell count with 80% lymphocytes in the absence of infection. Nonimmune hydrops is reviewed in more detail in Chapter 123 .
There is a strong association between CH or other lymphedemas and genetic abnormalities, major congenital anomalies, perinatal loss, and other adverse outcomes. These abnormalities include genetic syndromes such as Angelman syndrome, Noonan syndrome, skeletal dysplasia, cystic fibrosis, neurofibromatosis, and cerebromandibular syndrome. Turner syndrome is the most common associated chromosomal abnormality, affecting approximately 60% of aneuploidies. In addition, more recent studies have shown a considerable risk for Down syndrome, accounting for about 30% of aneuploidies. Even fetuses with normal karyotype are at risk for major congenital anomalies; the majority are cardiac anomalies, and urinary, central nervous system, and body wall anomalies ( Table 121.1 ).
| Complications of Cystic Hygroma With Normal Karyotype | Defect Types |
|---|---|
| Cardiac anomaly | Hypoplastic left heart defect, VSD, aortic coarctation, tetralogy of Fallot |
| Genitourinary | Hydronephrosis, bladder malformations, hypoplastic or multicystic dysplastic kidney |
| CNS | Dandy-Walker malformation |
| Body wall defects | Gastroschisis, omphalocele, limb–body. wall complications |
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


