- •
Position of the conal septum in relation to the ventricular septum and size of the ventricular septal defect created
- •
Degree of posterior deviation of the conal septum and size of the left ventricular outflow tract
- •
Aortic annulus diameter and presence or absence of aortic valvar stenosis
- •
Size of the left ventricle (typically normal)
- •
Mitral valve size and anatomy (typically normal)
- •
Size and caliber of the ascending aorta, transverse aorta, and aortic isthmus
- •
Presence or absence of coarctation of the aorta or interruption of the aortic arch
- •
Identification of the branching of the aortic arch and the origin of the upper extremity and head vessels
- •
Identification of the presence or absence of thymic tissue in the anterior mediastinum
Anatomy and Anatomical Associations
The conal or infundibular septum is the superior-anterior portion of the ventricular septum. It separates the aortic and pulmonary outlets. Abnormal development can result when there is deviation of the conal septum posteriorly in relation to the remainder of the muscular septum. This posterior malalignment situates the conal septum across the left ventricular outflow tract (LVOT). A ventricular septal defect (VSD) coexists with this posterior malalignment ( Figure 13-1 ). This VSD can be located in the membranous, outlet, or muscular septum and is referred to as a posterior malalignment ventricular septal defect (PMVSD). Such VSDs are almost always large and do not undergo spontaneous closure. In hearts with normally related great vessels, the VSD can be related more to the pulmonary artery and some degree of override of the pulmonary artery over the ventricular septum can be seen.
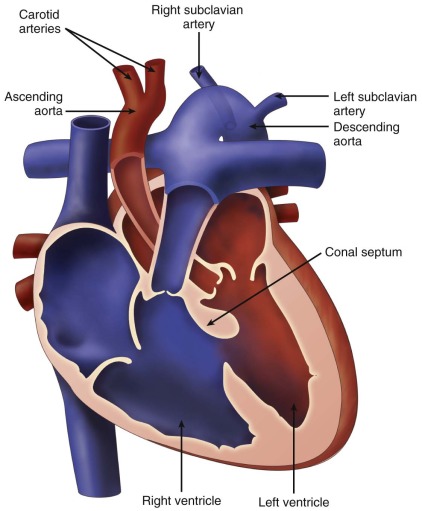
Depending on the degree of deviation of the conal septum into the LVOT, a PMVSD can be associated with narrowing of the subaortic region and decreased growth of the aortic valve. Thus, obstruction can be noted at the subvalvar or valvar level. In rare cases, posterior malalignment is associated with aortic atresia. In addition, a PMVSD can be associated with various degrees of aortic arch obstruction, including aortic arch hypoplasia, coarctation of the aorta, and interrupted aortic arch (IAA).
IAA is a complete absence of the connection between the ascending and the descending aorta. The classification of IAA is as follows, working from the distal end to the proximal end of the aortic arch :
- •
Type A: the interruption is distal to the left subclavian artery.
- •
Type B: the interruption is between the left common carotid and the left subclavian arteries.
- •
Type C: the interruption is between the innominate and the left common carotid arteries.
Type A interruptions can be associated with aortopulmonary window and type C interruptions are extremely rare.
Type B interruptions are more common than type A and are commonly associated with a PMVSD and subaortic obstruction. Bicommissural aortic valve is often seen with these defects. Arch sidedness and arch vessel abnormalities such as right aortic arch with aberrant right subclavian artery can also be associated with PMVSD with or without aortic arch obstruction and marks a higher probability of a deletion in chromosome 22q11 in those patients.
Frequency, Genetics, and Development
PMVSD is more often associated with IAA (93%) than with coarctation of the aorta (47%). Twenty-four percent of patients with PMVSD have a normal aortic arch. The incidence of IAA is 0.019 per 1000 live births and accounts for 1% of all congenital heart disease.
Genetic factors have been linked to the development of PMVSD and type B IAA. Type B IAA is considered a conotruncal defect because of the abnormal ventricular septal development and abnormal aortic arch formation. As with other conotruncal defects, namely, tetralogy of Fallot and truncus arteriosus, IAA type B is found in patients with deletion of chromosome 22q11. In fact, deletions involving chromosome 22q11 are seen in 57% of patients with type B IAA and 33% of patients with PMVSD. Interestingly, 22q11 deletions are more common in patients with associated vascular anomalies such as right aortic arch, aberrant right or left subclavian artery, aorticopulmonary collaterals, or absent/discontinuous branch pulmonary arteries. DiGeorge, velocardiofacial, and conotruncal anomaly face syndromes all involve chromosome 22q11 deletions. Extracardiac manifestations of these syndromes include hypoplasia/aplasia of the thymus and/or parathyroid glands, hypocalcemia, palatal abnormalities, learning disabilities, and facial dysmorphism. The strong association of PMVSD and IAA with this chromosomal abnormality lends evidence for the genetic origin of abnormalities involving the infundibular septum and conotruncus. The significantly increased prevalence of chromosome 22q11 deletion in IAA type B indicates that this particular cardiac defect has a different developmental origin from the other types of interruptions and from simple coarctation of the aorta.
Embryological factors may also explain the development of PMVSD and IAA. The branchial arterial system in the embryo undergoes significant change during development that involves regression of particular segments and growth of other segments into portions of the aorta and pulmonary arteries. In IAA of all types, certain parts of the branchial arch system involute and the corresponding arch segment fails to form.
Lastly, reduced blood flow to the aorta may explain the development of aortic arch obstruction. PMVSD can create significant obstruction that reduces blood flow through the LVOT and aorta. Blood flow is directed preferentially to the pulmonary valve through the ductus arteriosus to the descending aorta, leaving the aortic arch and isthmus with reduced blood flow compared with normal. This reduced blood flow is thought to prevent normal growth of the aortic arch and produce various forms of obstruction.
The heterogeneity of aortic arch obstruction and the variety of intracardiac defects make it difficult to propose a unifying etiology for PMVSD and aortic arch obstruction. For instance, greater than 90% of patients with IAA type B have PMVSD versus 50% of patients with type A interruption. Therefore, there may be a difference in etiologies between these subtypes. Genetic differences make it more likely that IAA type B is due to a primary developmental defect rather than a flow-related etiology related to the PMVSD. It is important to take these developmental, genetic, and physiological factors into consideration when evaluating the fetus and counseling families regarding these particular congenital heart abnormalities.
Fetal Physiology
Significant LVOT obstruction in the presence of a PMVSD reduces the blood flow across the aortic valve. Reduced forward flow across the aortic valve then reduces the percentage of combined cardiac output that traverses the aortic arch and isthmus. The majority of the combined cardiac output is now directed across the pulmonary valve to the ductus arteriosus and into the descending aorta. Because more left ventricular blood traverses the pulmonary valve, the oxygen content of the pulmonary arteries and of the descending aorta is higher than normal, but this change does not have a clinically noticeable impact on the development of the pulmonary vasculature or on lower body metabolism.
Because of the unrestrictive VSD, flow into the left ventricle (LV) is maintained. Therefore, unless there is mitral valve obstruction, LV growth continues and the LV is typically of normal size and is viable as a systemic ventricle. The right ventricle (RV) may be dilated due to the (1) increased flow from the VSD and (2) aortic arch obstruction.
In utero, flow across a VSD is usually bidirectional. An exclusive left-to-right shunt across the VSD suggests significant LVOT obstruction. In addition to alteration of flow across the VSD, LVOT obstruction may cause the shunt across the patent foramen ovale to be bidirectional. However, continuous left-to-right shunting across the foramen ovale is not expected and, if present, should raise concerns about the mitral valve or compliance of the LV.
Prenatal Management
Unlike other forms of aortic arch obstruction, the fetus with PMVSD and arch obstruction may not have LV-RV size discrepancy because of the associated VSD and ability for the ventricle to fill and have normal flow into it and out. Therefore, the standard four-chamber view is not likely to be abnormal unless there is mitral valve pathology or severe aortic valve hypoplasia/atresia. Angling anterior from the four-chamber view toward the LVOT will demonstrate the VSD, posterior malalignment of the infundibular septum, and encroachment of conal septum upon the subaortic region and the valve annulus. Assessment for the presence of subvalvar or valvar narrowing can be made with two-dimensional imaging. Doppler assessment may not detect LVOT obstruction in the presence of a large VSD with a left-to-right shunt. Angling further anteriorly will demonstrate the pulmonary valve and assessment of size discrepancy between the two outflow tracts can be made. The degree of pulmonary valve override of the VSD and the proportion of VSD shunt into the aorta versus the pulmonary artery can be determined. Angling further superiorly from the four-chamber view will visualize the great vessels in short axis and allows further assessment of size discrepancy between the great arteries. Normally, in this three-vessel view, the pulmonary artery is larger than the aorta and in turn, the aorta is larger than the right superior vena cava (RSVC). In the presence of a PMVSD and aortic arch obstruction, the aorta is significantly smaller than the pulmonary artery and the RSVC, which is easily demonstrated in the three-vessel view.
In the long-axis echocardiographic views, the LVOT, the degree of posterior malalignment, and the degree of LVOT narrowing are ideally assessed in an orthogonal plane to the four-chamber view. The direction of VSD shunt can be assessed in this plane where the color Doppler beam is parallel to flow. Normally, there is a bidirectional shunt across a VSD in utero. In the presence of LVOT obstruction, the shunt may be predominantly left to right. The ventricular septum should be scanned in its entirety by two-dimensional and color Doppler imaging to detect additional VSDs, keeping in mind that it may be difficult to detect additional VSDs in the setting of a large PMVSD.
Short-axis echocardiographic views of the heart can demonstrate a bicuspid aortic valve depending on the clarity of acoustic windows. The VSD is demonstrated in yet a different plane. A short-axis view of the mitral valve enables demonstration of structural abnormalities of the valve in the setting of mitral stenosis.
Intracardiac findings of LVOT obstruction, large VSD (especially in the presence of posterior malalignment of the conal septum), or aorta–pulmonary artery size discrepancy should prompt a full assessment of the aortic arch. Posterior malalignment of the conal septum should raise suspicion for arch hypoplasia, coarctation of the aorta, or type B IAA. A large mid-muscular VSD or LV-RV size discrepancy should raise the possibility of type A or C aortic interruption. Aortopulmonary window can be seen with type C interruption.
A long-axis view of the aortic arch will demonstrate the site of the interruption. In type B interruption (between the carotid and the subclavian arteries), the ascending aorta travels superiorly (or cranially) and does not arch back posteriorly. The subclavian artery is usually seen arising from the descending aorta just past the ductal insertion. It is easy to mistake the ductal arch for an aortic arch. Therefore, care must be taken to confirm that head and neck vessels arise from the arch before identifying it as the aortic arch versus the ductal arch. Arch sidedness may be assessed by angling superiorly from the three-vessel view. If the ductal and/or aortic arch connects to the descending aorta to the right of the spine, it is a right aortic arch. Arch-sidedness abnormalities, arch vessel anomalies, and the absence of thymic tissue should heighten suspicion for deletion of chromosome 22q11.
It is important to obtain serial fetal cardiology evaluations throughout the pregnancy because LVOT obstruction can be progressive over time. Parental counseling should include a discussion of possible genetic syndromes, including 22q11 deletion. Chorionic villous sampling or amniocentesis is recommended and important because an associated genetic syndrome can alter the prognosis of these cardiac defects. If there is ductal-dependent circulation, plans must be made to deliver in a center that can administer prostaglandin at birth to maintain ductal patency until a definitive neonatal surgical intervention can be made.
The essential question in PMVSD and arch obstruction is whether or not the LVOT will have the potential to accommodate a complete cardiac output across it. Hence, serial measurement of the narrowest portion of the LVOT in the long-axis view of the heart is important. Currently, there are no specific guidelines for measurements at different gestational ages that can predict one way or the other for adequacy or inadequacy of the size of the LVOT. Nevertheless, we have found that at our center, full-term neonates of at least 3 kg will do well with closure of VSD and arch repair if they have a measured LVOT of at least 4 mm in diameter. We have extrapolated from this experience to say that the fetus diagnosed in the second trimester with LVOT measuring at least 3 mm will likely have a bit more growth and have an adequate-sized LVOT at birth. Conversely, the presence of an LVOT measured in the third trimester that is less than 3 mm in diameter may reflect an inadequate LVOT for which an alternate surgical strategy beyond VSD closure and arch repair should be employed (see later).
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


