Abstract
Megacystis microcolon intestinal hypoperistalsis syndrome (MMIHS) is also called neonatal hollow visceral myopathy or Berdon syndrome . MMIHS is characterized by functional intestinal obstruction, dilated small intestine, microcolon, malrotation, and a massively enlarged bladder with or without megaureter and hydronephrosis but without lower urinay tract obstruction. With 227 reported cases before 2011, it is a rare condition mainly found in female fetuses and neonates. A mutation in the ACTG2-gene coding for smooth muscle gamma-2 actin is present in a relevant number of cases. Relations to intestinal smooth muscle myopathy, changes in the cells of the myenteric plexus, and dysfunctional nicotinergic acetylcholine receptors have been described.
Prenatally, a distended urinary bladder with normal or increased amniotic fluid; a dilated stomach, esophagus, and duodenum; segmental colonic dilation; and anomalies of the abdominal wall can be found sonografically or in magnetic resonance imaging studies. Postnatally, abdominal distension, absent bowel sounds, bile-stained vomiting, failure to pass meconium, and palpably enlarged kidneys are findings in affected children. Therapeutic attempts include transabdominal drainage of the urinary bladder to prevent secondary damage to the proximal urinary system, total parenteral nutrition, use of prokinetic agents, and surgical correction of malrotation. Multiorgan transplantation has been conducted in single cases. Prognosis is poor, with most affected children dying of multiorgan failure after sepsis within the first months of their lives. There are occasional reports of individual survival up to 24 years of age.
Keywords
megacystis microcolon intestinal hypoperistalsis syndrome, megacystis, microcolon, intestinal malrotation, neonatal hollow visceral myopathy, ACTG2, smooth muscle gamma 2 actin, LUTO, prune-belly syndrome
Introduction
Megacystis-microcolon-intestinal hypoperistalsis syndrome (MMIHS) is a rare and severe condition that is characterized by functional intestinal obstruction with dilated small intestine, microcolon, malrotation, decreased or absent bowel movement, and, prenatally, a massively enlarged nonobstructive urinary bladder often associated with hydroureteronephrosis. The anomaly was first described by Berdon et al. in 1976 and is also termed neonatal hollow visceral myopathy . Female fetuses are affected four to five times more often than male fetuses. In the neonate, the predominant problem is the reduced or absent bowel function and the impossibility of enteral nutrition. Kidneys and the lower urinary tract are not dysfunctional in most cases; however, secondary impairment of the urinary system owing to urethral reflux is frequent.
The usual postnatal therapeutic approaches are total parenteral nutrition combined with a trial of various prokinetic agents (which remain ineffective in most cases). Transabdominal drainage of the urinary bladder is frequently applied to preserve kidney function. Further steps are laparotomy, attempts to milk out or resect dysfunctional parts of the intestine, or drain them via ileostomy. Multiorgan transplantation is a desperate therapeutic effort to provide a viable intestine in these children. Although most authors describe MMIHS as a fatal condition with newborns surviving for only a few months with all therapeutic efforts being made, there are reports of prolonged survival up to the age of 18 years.
Disease
Definition
MMIHS is defined by the presence of a massively distended urinary bladder without lower urinary tract obstruction, a dilated small bowel, and microcolon with decreased or absent peristalsis.
Prevalence and Epidemiology
From the first description in 1976 until 2011, more than 227 cases were reported. Females are affected more often than males. Although a possible autosomal recessive inheritance pattern has been suggested based on cases with a family history or consanguinity, most cases seem to be sporadic. Whole-exome sequencing recently showed that a relevant proportion of MMIHS is caused by de novo heterozygous mutations of the gene encoding the smooth muscle gamma-2 actin, ACTG2. In this case, a recurrence risk would be approximately 1%, while in the majority of familial disease an autosomal dominant inheritance pattern has been postulated.
Moreover, these studies show that the different clinical manifestations depend on the degree of protein malfunction and differential expression of smooth muscle actins.
Etiology and Pathophysiology
Recent findings suggest that the loss of intestinal function is related to intestinal smooth muscle myopathy. Reduction or absence of actin in the circular layer of the small intestine, and vacuolic degeneration of smooth muscle cells with reduction of contractile and cytoskeleton proteins, have also been described. A dysfunctional neuronal nicotinic acetylcholine receptor and an association with chromosome 15q11 deletion are being discussed. Earlier reports suggested various changes in ganglion cells and axons of the myenteric plexus as a reason for intestinal malfunction. An inflammatory process of the intestine and urinary tract, and defects in the synthesis of collagen fibers, are other hypotheses. Similar findings have been described in megacystis, megaureter, and fetal hydronephrosis.
Manifestations of Disease
Clinical Presentation
The diagnosis of MMIHS is usually made in neonates with functional intestinal and urinary tract obstruction. Clinical signs may be a distended abdomen, malrotation of the colon, and absent bowel sounds. Feeding intolerance and emesis may be found, and enlarged kidneys may be palpated. In contrast to megacystis secondary to urethral valve or other lower urinary tract obstructions (LUTO), a transurethral drainage of the bladder is possible. Cases resemble neuropathic bladder outlet obstruction, but without any form of meningomyelocele. Maldescent of the testes is common in affected males.
Abdominal surgery to correct malrotation, remove adhesions, and rinse the intestinal lumen via intraoperative catheterization of the intestine is frequently performed. Total parenteral nutrition is necessary in most cases, often followed by liver failure owing to chronic cholestasis. Because of the dysfunctional bladder, repeated urinary tract infections and eventual renal failure can occur. Affected children often die of multiorgan failure after sepsis in the first months of their lives.
Imaging Technique and Findings
Ultrasound.
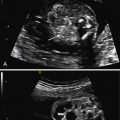
The leading sign of MMIHS is a massively distended bladder (megacystis) ( Fig. 30.1 ), combined with a normal or increased amount of amniotic fluid.

Additionally, mild bilateral hydronephrosis and isolated dilated stomach, or both, are often present. These signs may precede the development of megacystis and should be an indication for follow-up examinations. The bladder wall is usually not thickened. When fetal abdominal cysts are detected, MMIHS should be part of the differential diagnosis. Megacystis may develop only after 20 weeks’ gestation, in contrast to LUTO, in which megacystis is often detected in the first trimester ( Fig. 30.2 ). Because there is a predilection for female infants, knowledge of fetal gender may be valuable. Additional US findings may be a distended stomach ( Fig. 30.3 ), possibly in combination with a dilated duodenum and esophagus. Structural anomalies of the abdominal wall such as omphalocele, hydrometrocolpos, and segmental colonic dilatation can be additional features. In a recent review of prenatal diagnosed MMIHS cases, gastrointestinal abnormalities were noted in 24% of pregnancies complicated by MMIHS.

Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


