Metabolic and storage lung diseases are a broad group of diseases and syndromes characterized by underlying biochemical or metabolic dysfunctions. Accurate diagnosis is difficult because these diseases are often indolent, are rarely encountered in clinical practice, and because their manifestations are often vague and nonspecific. These groups of disorders may affect the lung solely or as part of a systemic disorder.
Metabolic Pulmonary Diseases
Pulmonary Alveolar Proteinosis
Etiology
Pulmonary alveolar proteinosis (alveolar lipoproteinosis) is a rare disease characterized by the accumulation of protein- and lipid-rich material resembling surfactant within the parenchymal airspaces. Three distinct subtypes of the disease are recognized: autoimmune or acquired, secondary, and genetic. More than 90% of cases are acquired and likely of autoimmune etiology related to a mutation in the surfactant gene or the presence of an antibody to granulocyte-macrophage colony-stimulating factor. These mutations and antibodies result in the defective clearance of surfactant by alveolar macrophages, as well as impaired function of lung neutrophils. Less commonly, pulmonary alveolar proteinosis is secondary, occurring in association with conditions that result in functional impairment or reduced numbers of alveolar macrophages, as may occur in inorganic dust inhalation, particularly acute silicosis; immunodeficiency syndromes, such as acquired immunodeficiency syndrome or immunoglobulin deficiency; and hematologic malignant neoplasms, such as acute myelogenous leukemia. Rarely, it may be congenital and rapidly fatal.
Prevalence and Epidemiology
The prevalence of pulmonary alveolar proteinosis has been estimated to be approximately 4 per million persons. Pulmonary alveolar proteinosis occurs predominantly in patients between the ages of 20 and 50 years and has a male-to-female preponderance of about 2.5 : 1. About 70% of patients are smokers.
Clinical Presentation
Patients may be asymptomatic in up to a third of all cases. The most frequent symptom is shortness of breath on exertion that is usually slowly progressive. Cough, usually nonproductive, is also common. A low-grade fever develops or is evident at initial evaluation in some patients and should suggest concomitant infection. Clubbing of the fingers has been identified in about a third of patients.
Pathophysiology
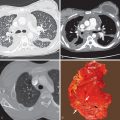
On histologic examination the alveoli can be seen to be filled with finely granular, lipoproteinaceous material that stains eosinophilic with hematoxylin and eosin and purple with periodic acid–Schiff ( Fig. 38.1 ). The alveolar architecture is usually preserved, but septal thickening may be due to edema, dilated lymphatics, or lymphocytic infiltration. Ultrastructural examination by transmission electron microscopy shows the intraalveolar material to consist of amorphous granular debris containing numerous relatively discrete osmiophilic granules or lamellar bodies. These structures represent phospholipids and are identical to inclusions found in normal type II pneumocytes.

Pulmonary function test results may be normal or show a restrictive ventilatory defect with reduction in vital capacity and total lung capacity and a disproportionately severe decrease in the carbon monoxide diffusing capacity (DLCO). Extensive disease may result in hypoxemia and increased alveolar-arterial pressure gradient (P ao 2 -Pa o 2 ), which increases further with exercise.
Manifestations of the Disease
Radiography
The characteristic radiographic pattern of pulmonary alveolar proteinosis consists of bilateral, symmetric patchy areas of consolidation that have a vaguely nodular appearance. In up to 50% of cases, the consolidation is perihilar (bat wing or butterfly distribution; Fig. 38.2 ); in the remaining cases, it is random or has a predominantly peripheral or basal distribution ( Fig. 38.3 ). In up to 84% of cases, there is regional sparing, most commonly the lung apices, costophrenic sulci, or lung periphery. Characteristically, patients may have extensive consolidation and relatively mild respiratory symptoms (“clinicoradiologic discrepancy”). In patients who have less severe disease, the appearance may be one of ground-glass opacities. Frequently, the parenchymal involvement is asymmetric or rarely unilateral. In some patients a linear interstitial pattern can be seen superimposed on consolidation or ground-glass opacities.


High-Resolution Computed Tomography
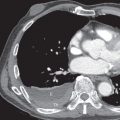
High-resolution computed tomography (HRCT) is superior to chest radiography in assessment of the pattern and distribution of abnormalities and may show lesions even when radiography is normal. The predominant abnormality typically consists of bilateral ground-glass opacities, although consolidation may also be present, particularly in the dorsal lung regions. The ground-glass opacities tend to involve all lung zones to a similar extent but can have a predominant lower zone distribution. In about 75% of cases in one series, a fine linear pattern forming polygonal shapes, measuring 3 to 10 mm in diameter, can be seen superimposed on the ground-glass opacities (“crazy paving” pattern; see Figs. 38.2 and 38.3 ). The pattern reflects the presence of interstitial edema or the accumulation of lipoproteinaceous material in the airspaces adjacent to the interlobular septa. There is typically sharp demarcation between normal and abnormal parenchyma. Pleural effusions and lymphadenopathy are always absent and should raise suspicion for an alternative diagnosis, secondary pulmonary alveolar proteinosis, or the possibility of coexistent infection. The radiologic manifestations of secondary pulmonary alveolar proteinosis are often similar to those of the more common idiopathic form ( Fig. 38.4 ). However, some subtypes, such as silicoproteinosis, may manifest with dependent consolidation, often containing foci of calcification, as well as diffuse solid centrilobular nodules and patchy ground-glass opacities. Affected patients also have calcified mediastinal and hilar lymph nodes, and the “crazy paving” pattern is usually absent.

In reviewing the radiographic and CT images in a patient with known pulmonary alveolar proteinosis, it is important also to look for complications. The main complication is infection, including community-acquired pneumonia and infection caused by unusual organisms such as Nocardia, Aspergillus, and Pneumocystis . The increased susceptibility to pulmonary infection may be due to impaired macrophage and neutrophil function, as well as the presence of intraalveolar lipoproteinaceous fluid that may facilitate the growth of microorganisms. The prevalence of opportunistic infections has decreased considerably since the use of bronchoalveolar lavage (BAL) for the treatment of these patients.
Interstitial fibrosis occasionally develops in patients with pulmonary alveolar proteinosis manifesting as airway irregularity and fissural displacement ( Fig. 38.5 ). Other rare complications include the development of emphysematous bullae and pneumothorax. Pulmonary alveolar proteinosis has been shown to demonstrate increased fluorodeoxyglucose uptake, which is thought to be related to glucose use by the inflammatory components.

Differential Diagnosis
The findings on the chest radiograph mimic those of pneumonia, pulmonary edema, and hemorrhage, but unlike those other conditions, a patient with pulmonary alveolar proteinosis is much less symptomatic than the radiographic appearance would suggest. The diagnosis of pulmonary alveolar proteinosis can often be suggested by the characteristic manifestations on HRCT. However, although it is characteristic of pulmonary alveolar proteinosis, the “crazy paving” pattern can also be seen in a variety of other conditions, including primary lung or metastatic adenocarcinoma, lipoid pneumonia, pulmonary hemorrhage or edema, and infections, including bacterial or Pneumocystis pneumonia. The diagnosis of pulmonary alveolar proteinosis can usually be confirmed by examination of BAL. Characteristic features of pulmonary alveolar proteinosis include milky BAL fluid; relatively few inflammatory cells, including alveolar macrophages; large acellular eosinophilic bodies in a diffuse background of granular basophilic material; periodic acid–Schiff staining of the proteinaceous material; elevated levels of surfactant proteins; and characteristic ultrastructural features with abundant lamellar bodies and cellular debris. The main constituent of the BAL fluid in pulmonary alveolar proteinosis is phospholipid, mainly lecithin, the main component of surfactant. If it is deemed necessary to confirm the diagnosis by tissue examination, transbronchial biopsy is likely to be sufficient.
Synopsis of Treatment Options
Treatment of acquired pulmonary alveolar proteinosis is with unilateral or bilateral whole-lung lavage. Some patients require only one or two procedures, whereas a few require that lavage be repeated semiannually or annually. The overall prognosis for alveolar proteinosis treated by whole-lung lavage is excellent. Corticosteroids should not be used as empirical treatment of alveolar proteinosis because of its potential to exacerbate opportunistic infections. Spontaneous resolution without treatment may occur in up to 25% of patients. On occasion, patients may not respond to BAL and may require lung transplantation or experimental therapy with granulocyte-macrophage colony-stimulating factor or rituximab. As might be expected in view of the pathogenesis, recurrence of the condition has been documented after lung transplantation.
Therapy for secondary pulmonary alveolar proteinosis is aimed at treatment of the underlying condition. For example, in patients with pulmonary alveolar proteinosis secondary to hematologic malignant disease, the pulmonary findings improve or resolve after successful chemotherapy or bone marrow transplantation.
- •
It is a rare disease, usually of autoimmune etiology.
- •
Lipoproteinaceous material (surfactant) fills the airspaces.
- •
Imaging findings:
- •
Ground-glass opacities or consolidation.
- •
Perihilar, peripheral, or random distribution.
- •
High-resolution CT usually shows a characteristic pattern of smooth interlobular septal thickening and intralobular lines superimposed on ground-glass opacities (“crazy paving” pattern).
- •
Abnormal lung is sharply demarcated from normal lung.
- •
Pleural effusion or lymphadenopathy is rare and should suggest an alternative diagnosis—secondary pulmonary alveolar proteinosis or coexistent infection.
- •
- •
Bronchoalveolar lavage is usually diagnostic.
Amyloidosis
Etiology
Amyloidosis is a generic term for a heterogeneous group of disorders characterized by accumulation of various insoluble fibrillar proteins (amyloid). The abnormal proteins are deposited in the extracellular space and cause disease by compression of the adjacent cells and tissues. Amyloidosis can be hereditary or acquired, localized, or systemic.
Prevalence and Epidemiology
Amyloidosis is rare. It affects the respiratory tract in about 50% of cases. Amyloid may involve the trachea, bronchi, mediastinum, pleura, heart, or, more commonly, the lung parenchyma.
Clinical Presentation
Tracheobronchial amyloidosis typically manifests after the fifth decade with dyspnea, cough, and occasionally hemoptysis. Less commonly, it causes symptoms that simulate asthma. Discrete tracheal and endobronchial nodules are usually asymptomatic but occasionally may result in airway obstruction with distal atelectasis or bronchiectasis. Symptoms and signs in such cases depend on the volume of lung affected and whether infection is present. The presence of amyloid in other sites is rare.
The nodular parenchymal form of amyloidosis usually is asymptomatic and is discovered incidentally on the chest radiograph. The majority of patients have no evidence of extrathoracic disease (either amyloidosis or otherwise).
Diffuse interstitial amyloidosis frequently results in progressive dyspnea and respiratory insufficiency. Diffuse involvement is most often seen as part of multisystem disease (primary amyloidosis), in which amyloid type L is typically present and may be associated with multiple myeloma.
Pathophysiology
Amyloidosis is a disorder of protein folding rather than of amino-acid sequence, and amyloid deposits consist mainly of protein fibrils. Amyloid is not a single substance but consists of several proteins, each of which resembles the others morphologically but is distinctive biochemically. The most important proteins associated with respiratory tract disease are amyloid L and amyloid A. Amyloid L is derived from immunoglobulin light chains and is therefore usually associated with abnormal plasma cell function, either as part of a systemic disease, such as multiple myeloma or macroglobulinemia, or, more commonly, localized to the lungs without evidence of systemic disease. Amyloid A is derived from a serum acute-phase reactant synthesized in the liver that can be formed in several chronic inflammatory diseases, including connective tissue disease, particularly rheumatoid arthritis; chronic infection, particularly tuberculosis; bronchiectasis; and certain neoplasms, such as Hodgkin disease. The majority of cases of respiratory tract amyloidosis are amyloid type L; the main exceptions are in patients with chronic inflammatory disease or a family history of amyloidosis. On histologic examination, amyloidosis is characterized by the presence of amorphous, eosinophilic, extracellular material that shows a characteristic apple-green birefringence when it is stained with Congo red and examined by polarizing microscopy ( Fig. 38.6 ).

There are three major forms of amyloidosis in the lower respiratory tract: tracheobronchial, nodular parenchymal, and diffuse parenchymal (alveolar septal, interstitial). Although these forms can occur in combination, in many cases the amyloid is deposited predominantly at one site. In addition to airway and pulmonary parenchymal disease, amyloidosis can also affect the hilar and mediastinal lymph nodes, pulmonary arteries, pleura, heart, and diaphragm.
Airway involvement occurs most commonly in the trachea and proximal bronchi. Although overlap does occur, it is usually manifested in one of two ways: a localized nodule or, more commonly, multiple discrete or confluent intramural plaques that distort the airway wall and cause stenosis of its lumen. On histologic examination the amyloid is situated in the subepithelial interstitial tissue and often surrounds tracheobronchial gland ducts and acini.
The parenchymal nodules of localized pulmonary amyloid can be solitary or multiple and are usually fairly well defined. Amyloid is often identifiable in the alveolar interstitium at the periphery of the nodule; however, in the central region the normal parenchymal architecture is generally obscured by a more or less solid mass of amyloid that typically contains fairly numerous multinucleated giant cells and variable numbers of lymphocytes and plasma cells. Calcification and ossification are relatively common. The nodular parenchymal form of amyloidosis is usually of amyloid type L and localized to the lung. It is the most common respiratory form of amyloidosis. In one series of 48 patients with localized respiratory tract amyloidosis, 34 had parenchymal deposits, of which 28 cases were nodular and 6 were diffuse alveolar septal.
Diffuse interstitial (alveolar septal) amyloidosis involves the parenchymal interstitium and the media of small blood vessels. The amyloid deposition is typically adjacent to endothelial and epithelial basement membranes and can appear in a uniform and more or less linear pattern or as multiple small nodules.
Lung Function
Pulmonary function tests may show evidence of restriction and impaired gas transfer in patients who have diffuse alveolar septal amyloidosis and air-trapping and fixed upper airway obstruction in those who have proximal tracheobronchial involvement.
Manifestations of the Disease
Radiography
In the majority of patients with tracheobronchial amyloidosis, the chest radiograph is normal. When present, the findings include focal or diffuse thickening of the airway wall with associated narrowing of the airway lumen ( Fig. 38.7 ). The findings are often difficult to see on the radiograph.

Nodular primary parenchymal amyloidosis manifests as solitary or, less commonly, multiple nodules usually ranging from 0.5 to 15 cm in diameter. The nodules occur most commonly in the lower lobes and are usually peripheral. Disease may progress slowly during a period of several years, with a slight increase in size of the nodules and the development of additional nodules.
Diffuse interstitial (alveolar septal) amyloidosis results in a reticular, nodular, or reticulonodular pattern that may be diffuse or involve mainly the lower lobes.
Pleural effusions may occur in amyloidosis but are usually due to heart failure caused by cardiac involvement. Pleural amyloid deposits are rare.
Computed Tomography
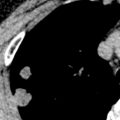
The CT manifestations of tracheobronchial amyloidosis consist of thickening of the airway wall, narrowing of the lumen, and, in some cases, foci of calcification ( Fig. 38.8 ; see Fig. 38.7 ). The airway wall thickening may be focal or diffuse and nodular, plaque-like, or circumferential; it is generally confined to the trachea but can extend to the main, lobar, and segmental bronchi. Bronchial involvement is frequently associated with distal atelectasis, bronchiectasis, or air-trapping.

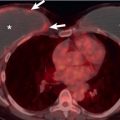
Nodular primary parenchymal amyloidosis manifests as solitary or, less commonly, multiple nodules usually ranging from 0.5 to 5 cm in diameter ( Fig. 38.9 ). On occasion, nodular parenchymal amyloidosis may result in a large mass ( Fig. 38.10 ). Calcification is seldom evident on radiographs but is seen in 20% to 50% of nodules on CT. The nodules tend to be more common in the periphery of the lower lobes but may be seen anywhere in the parenchyma (see Fig. 38.9 ). Rarely, nodules will cavitate. Cysts may occasionally be present adjacent to the nodules. Cysts and nodular amyloid deposits have been described most commonly in patients with Sjögren syndrome, with or without associated lymphoid interstitial pneumonia ( Fig. 38.11 ).



The HRCT findings of diffuse interstitial amyloidosis consist mainly of reticular opacities and interlobular septal thickening. Multiple subpleural micronodules can often be seen in association with the reticular opacities. Less common HRCT findings include ground-glass opacities, consolidation, traction bronchiectasis, and honeycombing. Punctate calcification may be seen in some of the nodules and areas of consolidation.
Mediastinal and hilar lymphadenopathy is rare in localized disease but occurs in approximately 75% of patients with amyloid L-type systemic amyloidosis ( Fig. 38.12 ). It is seen most commonly with diffuse interstitial amyloidosis.

Differential Diagnosis
The radiologic and CT findings are relatively nonspecific. The differential diagnosis of tracheal wall thickening includes relapsing polychondritis, tracheobronchopathia osteochondroplastica (TBO), granulomatosis with polyangiitis (formerly Wegener granulomatosis), inflammatory bowel disease, and adenoid cystic carcinoma. Unlike amyloidosis, both relapsing polychondritis and TBO typically spare the noncartilaginous posterior membrane. The main differential diagnosis of the parenchymal nodular form of amyloidosis is granulomatous infection or primary or metastatic tumors. The differential diagnosis of the diffuse interstitial form includes a large number of interstitial and airspace lung diseases, such as nonspecific interstitial pneumonia, usual interstitial pneumonia, and primary lung and metastatic adenocarcinoma. The diagnosis of amyloidosis usually requires histologic confirmation by needle, bronchial, transbronchial, or surgical biopsy. The diagnosis is based on demonstration of amyloid by Congo red staining, which produces characteristic apple-green birefringence under crossed polarized light.
Synopsis of Treatment Options
The treatment of symptomatic tracheobronchial amyloidosis may involve intermittent bronchoscopic resection, surgical resection, or laser ablation. Repeated bronchoscopic intervention is considered preferable to surgical resection, but recurrence is common. Stenting may have a role in the treatment of airway narrowing and complications thereof.
Nodular parenchymal amyloidosis is usually asymptomatic and remains stationary or progresses slowly. It usually has a good prognosis and seldom requires treatment. By contrast, diffuse interstitial amyloidosis frequently progresses. Treatment includes oral melphalan and prednisolone or peripheral stem cell transplantation. The natural history and prognosis of disease in patients who have diffuse interstitial pulmonary involvement may be determined by the presence of amyloid elsewhere in the body, particularly the heart and kidneys; however, many patients die of respiratory failure. In one series of 35 patients, the median survival time after diagnosis was only 16 months.
- •
Abnormal protein is deposited in extracellular tissues.
- •
Amyloidosis is a disorder of protein folding rather than of amino-acid sequence.
- •
The three main manifestations are tracheobronchial, parenchymal nodular, and diffuse interstitial.
- •
Tracheobronchial amyloidosis
- •
Clinical symptoms: dyspnea and cough; focal or diffuse thickening of the trachea or central bronchi that involves the posterior membrane
- •
Foci of calcification common
- •
Atelectasis and obstructive pneumonitis
- •
- •
Nodular parenchymal amyloidosis
- •
Usually asymptomatic
- •
Single or multiple nodules or masses
- •
Mainly lower lobe and peripheral location
- •
Foci of calcification common
- •
- •
Diffuse interstitial amyloidosis
- •
Clinical symptoms: progressive shortness of breath, dry cough
- •
Reticular pattern
- •
Ground-glass opacities on high-resolution CT (HRCT)
- •
Interlobular septal thickening on HRCT
- •
May be associated with lymphadenopathy and cardiac involvement
- •
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


