Considering the complexity of congenital midline abnormalities of the brain, it is mandatory to review and summarize the subject, based on embryologic development, developmental biology, clinical approach, neuroimaging, and molecular genetics. There are still several areas of poor knowledge, confusion, and controversy. Pathogenesis in some cases is not clearly understood, and definitions and limits among different diseases are not apparent. This article reviews several malformations of the skull and brain with the aims of making image interpretation easier, enabling an accurate diagnosis, establishing the prognosis, and aiding in genetic counseling.
Considering the complexity of congenital midline abnormalities of the brain, it is mandatory to review and summarize the subject, based on embryologic development. There are still several areas of poor knowledge, confusion, and controversy. Pathogenesis in some cases is not clearly understood, and definitions and limits among different diseases also unclear. Reviewing these concepts makes image interpretation easier, giving an accurate diagnosis that can help the clinician establish the prognosis and provide genetic counseling. Brain malformations are frequently diagnosed either incidentally or because of patients’ overall clinical picture. In some cases the radiologist does not know relevant data from the medical chart, such as the prenatal or family history, which thus can lead to a diagnosis based only on the imaging findings. The latter situation advocates a review of congenital midline abnormalities of the brain in order to clarify concepts and allow a diagnosis that is better oriented. Some investigators believe that a classification system based solely on neuroimaging criteria is not achievable.
Midline cephaloceles
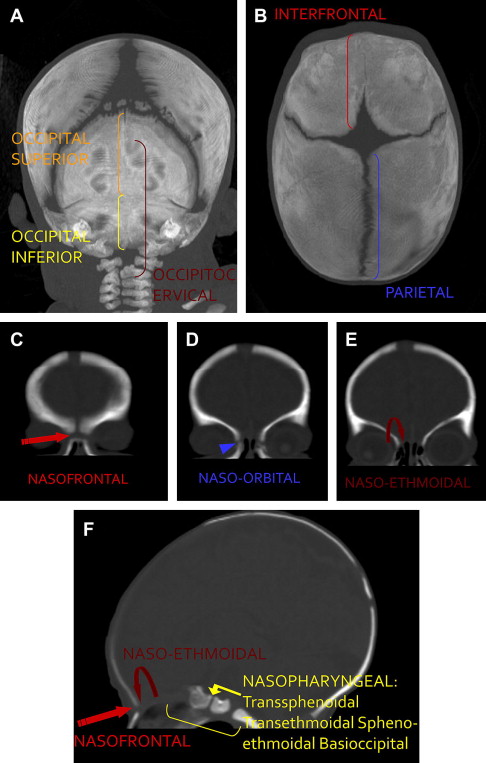
Cephaloceles are an extracranial protrusion of the cerebral tissue and meninges through a congenital defect in the cranial bones. The connection between the protruding tissue and the intracranial cavity remains. Cephaloceles are usually classified according to the location of the skull defect: anterior or posterior in relation to the coronal sutures ( Fig. 1 ). Anterior cephaloceles comprise those whereby the swelling is in front of the coronal suture or located in the orbit or nasal cavity; these include interfrontal, frontoethmoidal (nasofrontal, nasoethmoidal, naso-orbital), and nasopharyngeal (transsphenoidal, transethmoidal, sphenoethmoidal, basioccipital) cephaloceles. Posterior cephaloceles are defined as parietal when the defect is between the bregma and the lambda, and occipital when the defect arises between the lambda and the foramen magnum, possibly including one or more of the cervical vertebrae.
Atretic cephaloceles are formes fruste of cephaloceles, which contain a small, noncystic, flat, or nodular lesion. This lesion is situated in the midline of the scalp, either near the vertex (parietal) or just cephalic to the external occipital protuberance (occipital form). Other definitions are listed in Box 1 .
- •
Cranium bifidum occultum: a midline or paired paramedian skull defect, without the prolapsing of meninges or brain (eg, parietal or frontal foramina)
- •
Exencephaly: acrania with protrusion of the central nervous system (CNS) into the amniotic cavity (in utero) or into the environment (postnatal)
- •
Anencephaly: acrania with absence of most or all of the brain tissue
- •
Cutis aplasia congenita: absence of skin in a localized or extended area at birth. Variable depth of the defect (only epidermis or as deep as the dura)
Cephaloceles can also be classified according to the involvement of the cranial vault (interfrontal, parietal, occipital, and lateral) or the skull base (temporal, frontoethmoidal, nasopharyngeal, spheno-orbital, and sphenomaxillary). Nasopharyngeal, temporal and spheno-orbital types are classified as basal cephaloceles by other investigators. A detailed histologic examination allows for another classification according to their content: pure meninges (meningocele), meninges and brain (encephalomeningoceles), part of the ventricle (encephalocystocele), or glial-lined cyst containing cerebrospinal fluid (CSF) (gliocele).
The incidence of cephaloceles is approximately 0.8 to 3.0 per 10,000 births. Occipital cephaloceles constitute 66% to 89% of the cases, frontoethmoidal 15%, and basal 10%. Frontoethmoidal cephaloceles show a relatively high incidence (1:5000 live births) in Southeast Asia. There are several factors that influence the incidence of neural tube defects (NTD), such as different genetic alterations, nutrition and the health status of women, frequency of prenatal diagnosis resulting in an elective abortion, and exposure to NTD-inducing toxic agents.
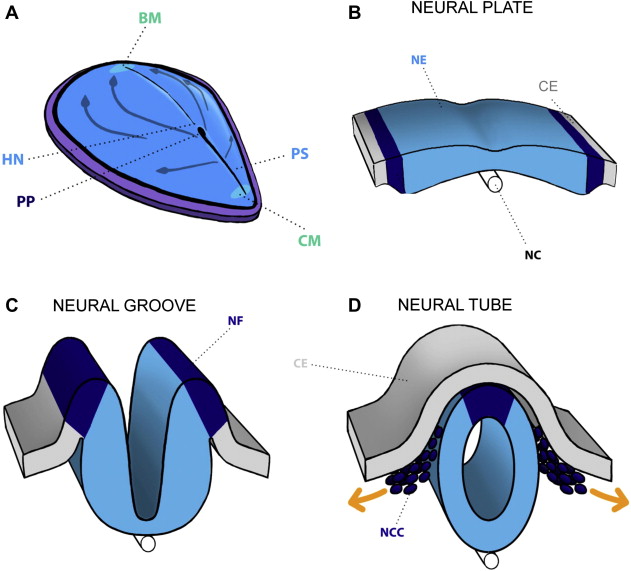
On the fifth day of life, gastrulation starts and establishes 3 germ layers (ectoderm, mesoderm, and endoderm) in the embryo. On days 15 to 16, invagination starts (cells enter Hensen’s node and migrate cephalad) to form the notochordal process, which eventually becomes the notochord. The notochord induces the overlying ectoderm to thicken and form the neural plate. Cells of the neural plate form the neuroectoderm, and their induction initiates the neurulation process. The expression of several genes and proteins that participate in the process of gastrulation and neurulation is described in Box 2 . At about 17 days gestational age (GA), the neural plate thickens laterally and forms the neural folds. These folds contract (due to the underlying actin myofibrils), elevate, and form a depressed medial zone known as the neural groove. At about the 20th gestational day, the folds thicken and the surrounding mesenchyme expands simultaneously, leading the edges of the folds to approach one another in the midline, where they fuse at the following levels: the mesencephalic-rhombencephalic boundary, the cervical spine, and the rostral end of the neural groove. As the folding edges of the neural ectoderm join, they are covered by adjacent cutaneous ectoderm. Disjunction of the two ectoderm (cutaneous and neural) types occurs as the edges meet, and the cells at the lateral border or crest of the neuroectoderm begin to dissociate from their neighbors, creating the neural crest ( Fig. 2 ). The neural crest cells migrate, and once they reach the prechordal skull base, the overlying neural tube is transformed into prosencephalon, which then differentiates into the diencephalon and telencephalon. Some of the neural crest derivatives include connective tissue and bones of the face and skull, cranial nerve ganglia, and dermis of the face and neck.
- 1.
Head-forming genes: OTX2, LIM1, HESX1, Secreted factor Cerberus
- 2.
Initiates and maintains integrity of node and primitive streak: gene Nodal (member of Transforming Growth Factor β family)
- 3.
Mesoderm ventralization during gastrulation: BMP-4 (Bone Morphogenetic Protein 4), requires the presence of FGF (Fibroblast Growth Factor secreted by node and primitive streak)
- 4.
Antagonize BMP-4 activity and dorsalize mesoderm to form the notochord and somitomeres in the head region, and induction of neurulation: chordin, noggin, follistatin
- 5.
Proteins of induction (inactivation of BMP-4) of neural plate structures (hindbrain and spinal cord): FGF, WNT3a
The mechanisms leading to the defective closure of the neural tube are still unknown. Recently, Trovato and colleagues proposed that NTD may be due to p53 downregulation or disruption, which diminishes the expression of hepatocyte growth factor/scatter factor (HGF), c-met, and STAT3 genes. Other high-risk factors associated with NTD are listed in Box 3 .
- 1.
Gene disorders:
- a.
Cerebrocostomandibular syndrome
- b.
Fraser syndrome
- c.
Meckel-Gruber syndrome
- d.
Waardenbur syndrome
- a.
- 2.
Physical agents:
- a.
X-irradiation
- b.
Hyperthermia
- c.
Stress
- a.
- 3.
Drugs:
- a.
Thalidomide
- b.
Folate antagonists
- c.
Androgenic hormones
- d.
Valproate
- e.
Carbamazepine
- f.
Hypervitaminosis A
- a.
- 4.
Substance abuse:
- a.
Alcohol
- a.
- 5.
Chemical agents:
- a.
Organic mercury
- b.
Lead
- a.
- 6.
Maternal infections:
- a.
Rubella
- b.
Cytomegalovirus
- c.
Toxoplasma gondii
- d.
Syphilis
- a.
- 7.
Maternal metabolic conditions:
- a.
Phenylketonuria
- b.
Diabetes mellitus
- c.
Endemic cretinism
- a.
There are two important embryologic clues in defining the pathogenesis of cephaloceles. First, there is a midline skull defect in congenital nasal cephaloceles, which corresponds with the sites of neural tube closure along the median (implies neurulation disorder). Second, the fact that a skin or an epithelial layer covers the cephaloceles suggests that the neural tube must have closed (implies postneurulation disorder).
There are 4 theories about the pathogenesis of cephaloceles, as follows:
- 1.
Neurulation disorder. Hoving postulated that a disturbance in the disjunction between the cutaneous and the neural ectoderm at the site of final closure of the rostral neuropore, possibly due to a lack of apoptosis, might result in a frontoethmoidal cephalocele. This process can be disturbed at any of the sites of neural tube closure, leading to the various cephaloceles as classified. Examples of these abnormal points of closure and cephaloceles are listed in Table 1 .
Table 1
Abnormal points of closure and cephaloceles
Point of Closure
Cephalocele
Foramen cecum (closure of frontal and ethmoid bones)
Between chiasmatic plate and the nasal fields
Frontoethmoid
Basal
Data from Hoving EW. Nasal encephaloceles. Childs Nerv Syst 2000;16(10–11):702–6.
- 2.
Postneurulation. At later stages of embryogenesis isolated focal cranial defects may arise, while the multiple ossification centers coalesce to allow the progressive ossification of the chondrocranium (from 35 days GA until after birth). Certain cephaloceles of the skull base could be secondary to failure of the correct coalescence of the ossification centers, leaving persistent dehiscence between two endochondral ossification centers.
- 3.
Defective tissular induction. A defective tissular induction that occurs at early embryogenesis is a plausible explanation of the concurrence of cephaloceles and both cerebral and craniofacial malformations. These malformations encounter prosencephaly, agenesis of the corpus callosum, anomalies of the visual and olfactory structures, and midline facial clefts for the anterior cephaloceles, and agenesis of the corpus callosum with dorsal cyst, callosal lipoma, and porencephaly for posterior cephaloceles. These abnormalities could present for anterior cephaloceles, at 28 to 35 days GA, during the induction of the mesoblast (by the notochord and the neural tube) to form the cranial base and facial structures. Blustajn and colleagues explain the association of transsphenoidal cephalocele and both ophthalmologic and endocrine anomalies by this mechanism. If there is no migration of the neural crest cells to the future prechordal skull base region, there is no appropriate induction of the overlying diencephalic neural tube during organogenesis of the optic nerves and the hypothalamic-pituitary axis. In the case of posterior cephaloceles, the defective induction and formation of the membranous cranial roof occur at 38 to 45 days GA. At later stages of embryogenesis, isolated focal cranial defects may arise, while the multiple ossification centers coalesce to allow the progressive ossification of the chondrocranium (from 35 days GA until after birth). Certain cephaloceles of the skull base could be secondary to failure of this process.
- 4.
Intracranial cyst. Diebler and Dulac also proposed another mechanism to explain the association of certain cystic intracranial malformations (such as Dandy-Walker cyst) with cephalocele. Diebler and Dulac mention that an increased intracranial pressure (due to intracranial mass or cyst) may lead to progressive thinning of the neuropil, which evolves into a focal defect in the cranial vault with cephalocele.
Swelling at the level of the cephalocele, bony defect, facial malformations (anterior cephaloceles), and intracranial malformations are certain features shared by patients with cephaloceles ( Table 2 ). Magnetic resonance (MR) imaging is the modality of choice for the evaluation of all of these abnormalities; even though computed tomography (CT) is more precise than MR imaging in delineating the margins and extensions of the bony defect.
| Abnormality | Cephalocele |
|---|---|
| Dysgenesis corpus callosum (DCC) | TSPH, N-ETH, N-FR, OCC, P, A-P |
| Arachnoid cyst associated to DCC | P, A-P |
| Hydrocephalus | I-FR, OCC, P |
| Optic nerve atrophy | OCC, N-PHR, FR-ETH |
| Dandy-Walker cyst, Chiari II malformation, microcephaly | OCC, P |
| Venous anomalies, prominent supracerebellar cistern | OCC, P |
| Cerebral and cerebellar migration anomalies | OCC |
| Chiari III | OCC |
| Interhemispheric lipoma | N-F |
| Proptosis | N-O |
It is necessary to determine the size, extent, and nature of the contents of the cephalocele. The size of the cephaloceles varies according to their location and type (tissue content within the sac).The dysgenesis of the corpus callosum and hydrocephalus are the most common associated intracranial abnormalities. Encephaloceles are isointense relative to gray matter with most MR imaging sequences, but they may be hyperintense on T2-weighted images because of gliosis. When cephaloceles contain meninges, they are isointense to CSF. If cephaloceles contain ventricle, in addition to the CSF signal intensity the walls of the ventricles are seen isointense to gray matter.
New techniques such as MR imaging and MR angiography provide precise information about venous sinuses and arteries. Kotil and colleagues have shown that 3-dimensional (3D) Fourier-transformation constructive interference in a steady state (CISS) sequence and T2-weighted reversed (T2R) imaging on 3 Tesla allow a superb demonstration of the intracranial venous anomalies associated with these cephaloceles. These anomalies include an anomalous upward course of the straight sinus (“vertical embryonic positioning of the straight sinus”), elongation of the vein of Galen, and splitting of the superior sagittal sinus. A prominent supracerebellar cistern has also been shown with these techniques. This evaluation is crucial for surgical planning, particularly of venous anomalies in occipital, parietal, and atretic parietal cephaloceles.
Occipital Cephalocele
The location of the bony defect in occipital cephaloceles may be infratentorial ( Fig. 3 ), supratentorial, or combined. These defects can also involve the posterior arch of C1 and C2 (occipitocervical cephalocele). The occipital cephalocele can be small or large (>50 mm), and is rarely larger in size than the head (giant cephalocele, Fig. 4 ) of the patient. The occipital encephalocele is part of the Chiari III malformation. The features of this malformation are described in detail in the section on posterior fossa malformations. Intracranial malformations and syndromes are associated with occipital cephaloceles, as listed in Box 4 .
- 1.
The fourth ventricle and the occipital horn may protrude into the cephalocele
- 2.
Associated intracranial malformations:
Cerebral and cerebellar migration anomalies (cortical dysplasia, gray matter heterotopias, microgyria of the medial temporal cortex, faulty convolution, inversion or absence of the dentate fascia and rudimentary to absent amygdala), cystic malformations of the posterior fossa, dorsal interhemispheric cyst, fusion of thalami, partial or complete absence of the anterior commissure, septum pellucidum, fornices, and mammillary bodies. Hydromyelia and a deformed spinal cord, optic nerve atrophy
- 3.
Associated syndromes: Chemke, Cryptophthalmos, Knobloch, Meckel, pseudo-Meckel, dyssegmental dwarfism, frontonasal dysplasia
Parietal and Atretic Parietal Cephaloceles
The bony defect in parietal cephaloceles more commonly lies near the posterior or anterior fontanelle. Under this bony defect there is a fenestrated falx or dural dehiscence, leading to the ostium and the interhemispheric fissure being widened. A tentorial cleft or fenestration is shown as a consequence of the insertion of the separated dural leaves (of the falx), which insert into the ipsilateral tentorial leaf ( Fig. 5 ). Some clinical findings of parietal cephaloceles and associated intracranial malformations are mentioned in Box 5 .
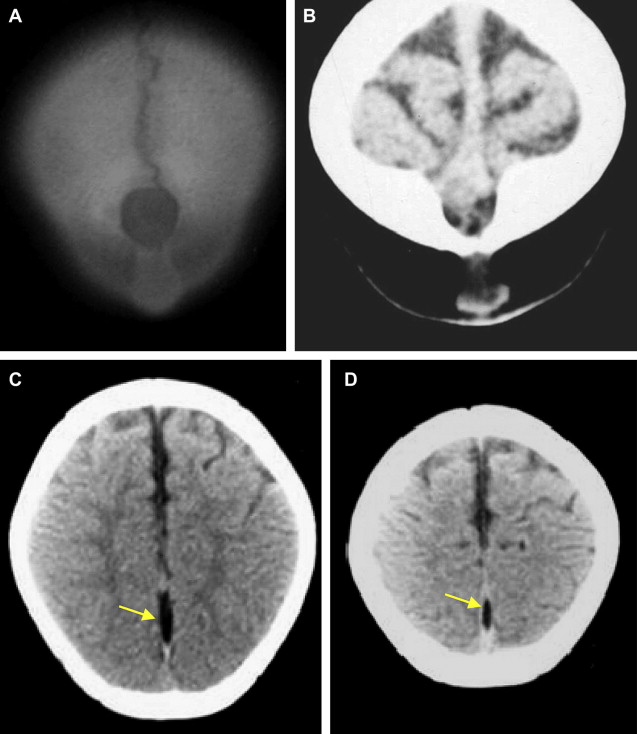
- 1.
Most are atretic (5–15 mm) and gliocele
- 2.
Microcephaly in 20%, marked retardation in 40%
- 3.
Typically display hair at the base and are rarely multiple
- 4.
Associated malformations: spectra of holoprosencephaly, vermian agenesis, arachnoid cyst with the dysgenesis of the corpus callosum, porencephalic cysts (atretic), and Walker-Warburg syndrome
Nowadays, thin-section (1–2 mm) gradient echo (with gradient spoiler) or fast spin echo MR images or thin CT images are available, and are required to visualize the small communication in atretic cephaloceles.
Anterior Cephaloceles
Midline anterior cephaloceles include (a) frontoethmoidal (nasoethmoidal, nasofrontal, and naso-orbital), (b) nasopharyngeal (transethmoidal, sphenoethmoidal, transsphenoidal, basioccipital), and (c) interfrontal.
Clinical manifestations of frontoethmoidal and nasopharyngeal cephaloceles may consist of nasal stuffiness, rhinorrhea, a broad nasal root, or hypertelorism. These cephaloceles change in size during crying, the Valsalva maneuver, or jugular compression (positive Furstenberg test). MR imaging evaluation must involve the specific zone and the entire brain. Associated anomalies with anterior cephaloceles are listed in Box 6 .
- 1.
Associated anomalies: arrhinencephaly, anophthalmia, microphthalmos, and dysgenesis of the corpus callosum, interhemispheric lipomas, arachnoid cyst, and hydrocephalus. Malformation of cerebral cortical development (schizencephaly)
- 2.
Associated syndrome: aberrant tissue band syndrome
- 3.
Interhemispheric lipoma in nasofrontal cephalocele
Frontoethmoidal (Sincipital) Cephaloceles
In frontoethmoidal cephaloceles, the internal skull defect is located in the midline, but the external skull defect may vary with the bony surroundings of the defect in the facial skeleton. Frontoethmoidal cephaloceles are soft tissue masses connected to the subarachnoid space via an enlarged foramen cecum, and extend to the glabella (nasofrontal) or into the nasal cavity (nasoethmoidal). A bifid or absent crista galli, cribriform plate, or a defect of the frontal bone suggest intracranial extension of the cephalocele, and are fairly well demonstrated with CT. It is important to bear in mind that no bone is seen in the normal neonate at the level of the crista galli and cribriform plate, because the crista galli and midline cribriform plate only start ossifying at 2 months (postnatal) and progress until about 14 months of age.
Nasoethmoidal (intranasal) cephaloceles are usually evident in the pediatric age group, and cause complex deformities in the frontal, orbital, and nasal regions. Sometimes it is a small defect and only manifests late in adulthood. In this setting, misdiagnosis is common as either a nasal polyp, intracerebral tumor that has invaded down through the skull base into the nasal anlage, and nasal gliomas. Biopsy is contraindicated in these cephaloceles because of potential CSF leaks, seizures, or meningitis.
In such cases, intrathecal injection of contrast material may show a subarachnoid connection in encephaloceles that is not seen in nasal gliomas. Cisternography with heavily T2-weighted thin sections may also be useful.
Hypertelorism is constantly seen in nasopharyngeal and nasofrontal cephaloceles, and commonly associates with nasoethmoidal and naso-orbital cephaloceles. Other facial malformations such as labial fissure, palatine fissure, and median nasal fissure are demonstrated in nasopharyngeal and frontoethmoidal cephaloceles ( Fig. 6 ).
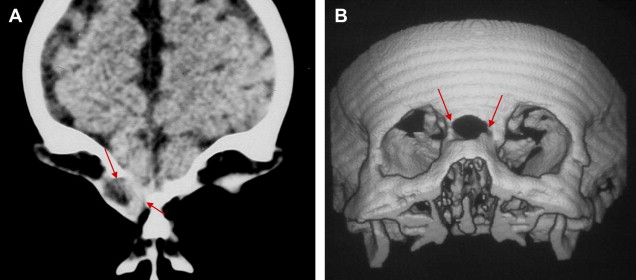
Because naso-orbital cephaloceles protrude into the orbit, they typically present proptosis and a slightly pulsatile mass.
Nasopharyngeal Cephaloceles
Nasopharyngeal cephaloceles are not obviously seen on clinical examination; because they are occult, they are discovered toward the end of the first postnatal decade. The cephalocele causes obstruction of the nasopharynx, which manifests as a persistent nasal stuffiness or “mouth-breathing.” Clinical examination detects a nasal or pharyngeal mass that increases in size with Valsalva maneuver, as mentioned earlier. It must be borne in mind that misdiagnosing nasopharyngeal cephaloceles as a mass might lead to an erroneous biopsy. To rule out a nasopharyngeal encephalocele, it is mandatory to perform a neuroradiological evaluation in patients with hypertelorism, diminished visual acuity and hypothalamic-pituitary dysfunction, and/or nasopharyngeal mass. These symptoms are due to the stretching of the inferior portion of the third ventricle, hypothalamus, and optic chiasm into the sac. Other ophthalmologic anomalies associated with these cephaloceles include hypoplasia of the optic discs, coloboma, and retinal dysplasia. Agenesis of the corpus callosum is demonstrated in approximately 80% of patients with nasopharyngeal cephaloceles.
Interfrontal Cephaloceles
Interfrontal cephaloceles are frequently located in the inferior part of the metopic suture; nevertheless, they may occupy the entire length of the suture, which allows the passage to the anterior tips of the frontal lobes or the anterior halves of the cerebral hemispheres. The protrusion of the brain could be asymmetric and extensive, leading to a rotation of the intracranial hemisphere. The associated cerebral malformations such as agyria, holotelencephaly (prosencephaly with arrhinencephaly), and hydrocephalus, and the compression of the herniated brain contribute to the poor prognosis of the interfrontal cephalocele ( Fig. 7 ).
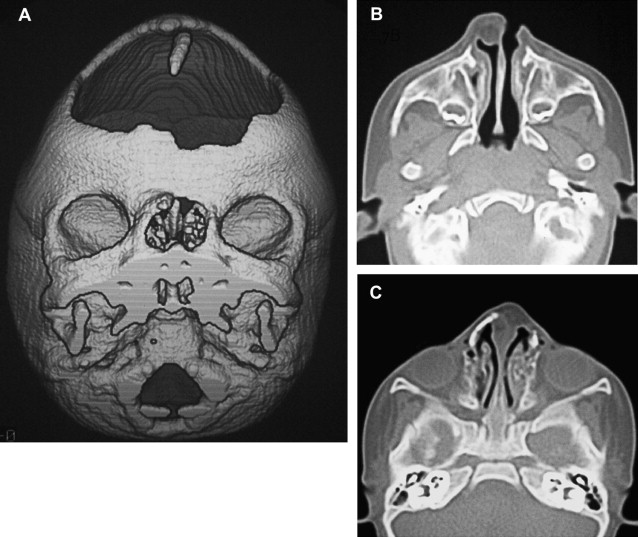
The treatment of congenital cephaloceles and CSF leaks includes craniotomies as well as, more recently, endoscopic surgery, and is aimed at performing the resection of the cephalocele and repairing the bony anomalies, particularly hypertelorism. This surgical resection must be performed at a very early age to prevent CSF leakage, meningitis, or increase in mass size. The prognosis of the patients with cephaloceles depends on its location, associated anomalies (both intracranial and extracranial), and size and content of the sac.
Nasal dermoids, nasal gliomas, and dermal sinus
The migration of neural crest cells forms centers of mesenchymal cells in the frontonasal region. There are spaces between these centers, prior to their fusion and ossification. The fonticulus frontalis is the space between the frontal and nasal bones, which will close postnatally and form the frontonasal suture. The prenasal space is the zone between the nasal bones and the precursor structures of the nasal septum and nasal cartilages. There is a dural diverticulum that extends through the prenasal space, reaches the tip of the nose, and regresses, allowing the closure of this space, which is then called the foramen cecum (situated anterior to the crista galli).
If brain tissue protrudes through the fonticulus frontalis and communicates with the intracranial cavity, a frontonasal cephalocele is formed. If the fonticulus frontalis closes after the protrusion of the brain tissue, the entrapped tissue constitutes a prenasal cerebral heterotopia (nasal glioma).
When the protrusion of the brain tissue is through the prenasal space without regression of this tissue, and connects to the intracranial cavity, a nasoethmoidal encephalocele arises ( Fig. 8 ).
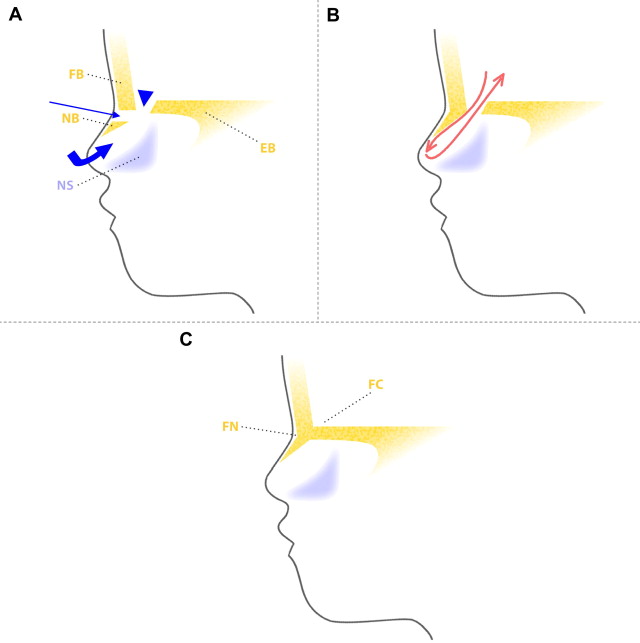
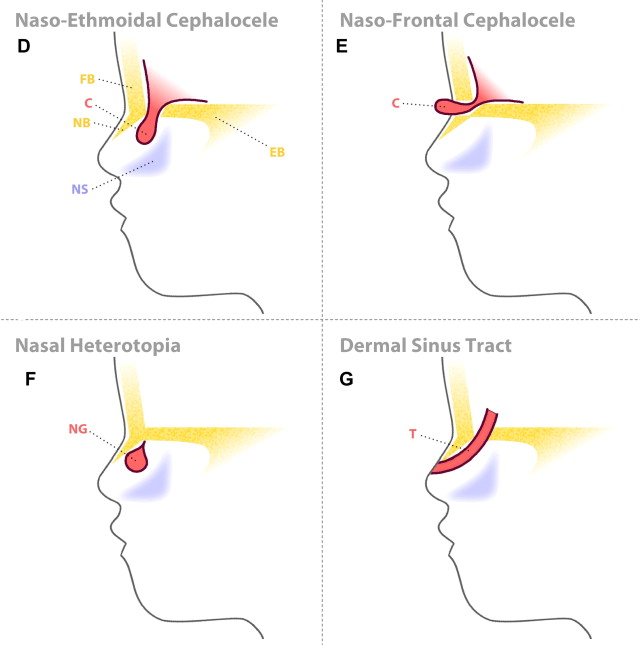
If dysplastic brain tissue remains separated from the intracranial CSF, after its protrusion through the prenasal space an intranasal cerebral heterotopia (intranasal glioma) is formed. There is a stalk of tissue in approximately 15% of cases, without direct fluid-filled tract connection with intracranial subarachnoid spaces. The partial regression of the most superior portion of the dural diverticulum is the postulated explanation for both the prenasal and the intranasal cerebral heterotopia.
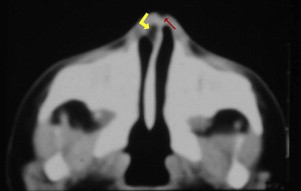
When the dural diverticulum reaches the dermis and remains attached (incomplete disjunction of the neuroectoderm and cutaneous ectoderm) to the skin in its regression through the prenasal space, a dermal sinus tract is created, leaving a dimple at its orifice. The dermal sinus tract may extend superiorly along the path of the dural diverticulum for variable distance, and can communicate with the intracranial cavity. Clinical findings of the dermal sinus tract include the presence of a dimple or pit ( Fig. 9 ) over the nasal bridge near the osteocartilaginous junction (or anywhere from the glabella to the tip of the nose), hairs that emanate from the pit, and sebaceous discharge. Some of the tissue within the sinus dermal tract can create dermoid and/or epidermoid cysts, due to persistent ectodermal elements at sites of suture closure, brain diverticulation, and neural tube closure. These cysts can also be found along the following midline locations: the anterior fontanelle, glabella, nasion, vertex, subocciput, at the level of foramen cecum (adjacent to the crista galli), and adjacent to the anterior margin of the third ventricle.
MR imaging is the best choice to achieve the objective sought in studying these lesions, namely, evaluating the nature (imaging features of the tissue), locating site of origin, and determining the extension of the lesion.
Nasal gliomas are evident near the root of the nose, located paramedian to the bridge of the nose and external to the nasal cavity, while intranasal gliomas are inside the nasal cavity medial to the middle turbinate bone.
MR imaging is the modality of choice for the evaluation of these lesions, which are hypointense to isointense relative to gray matter on T1-weighted and T2 -weighted imaging, with a focal high signal on T2-weighted sequences within the lesion. This latter finding is due to gliosis, cysts, and myxoid degeneration. Even though the dysplastic tissue does not enhance, the surrounding compressed mucosa does. To demonstrate the intracranial stalk, a high-resolution surface coil MR imaging method must be applied.
Concerning the dermal sinus tract, not only the features of the sinus tract but also the associated dermoid and epidermoid cysts must be detected. The dermal sinus tract is seen as a hypointense line on T1-weighted images within the subcutaneous tissue of the midline nasal bridge, extending toward the prenasal space.
The epidermoid cysts are isointense to CSF on both T1-weighted (low signal) and T2-weighted (bright signal) images, and show restriction on isotropic diffusion-weighted images.
Because dermoid cysts have fat components, they demonstrate T1 shortening (bright signal) and variable hyperintensity on T2-weighted images, as well as variable signal intensity on diffusion-weighted images.
A bifid anterior crista galli and an enlarged foramen cecum, plus atypical signal intensity at the foramen cecum, indicate the presence of an intracranial epidermoid or dermoid cyst.
All of the aforementioned lesions can be surgically resected. The nasal dermoid and epidermoid cysts are operated on to avoid potential CNS infection if there is intracranial connection.
Nasal dermoids, nasal gliomas, and dermal sinus
The migration of neural crest cells forms centers of mesenchymal cells in the frontonasal region. There are spaces between these centers, prior to their fusion and ossification. The fonticulus frontalis is the space between the frontal and nasal bones, which will close postnatally and form the frontonasal suture. The prenasal space is the zone between the nasal bones and the precursor structures of the nasal septum and nasal cartilages. There is a dural diverticulum that extends through the prenasal space, reaches the tip of the nose, and regresses, allowing the closure of this space, which is then called the foramen cecum (situated anterior to the crista galli).
If brain tissue protrudes through the fonticulus frontalis and communicates with the intracranial cavity, a frontonasal cephalocele is formed. If the fonticulus frontalis closes after the protrusion of the brain tissue, the entrapped tissue constitutes a prenasal cerebral heterotopia (nasal glioma).
When the protrusion of the brain tissue is through the prenasal space without regression of this tissue, and connects to the intracranial cavity, a nasoethmoidal encephalocele arises ( Fig. 8 ).
If dysplastic brain tissue remains separated from the intracranial CSF, after its protrusion through the prenasal space an intranasal cerebral heterotopia (intranasal glioma) is formed. There is a stalk of tissue in approximately 15% of cases, without direct fluid-filled tract connection with intracranial subarachnoid spaces. The partial regression of the most superior portion of the dural diverticulum is the postulated explanation for both the prenasal and the intranasal cerebral heterotopia.
When the dural diverticulum reaches the dermis and remains attached (incomplete disjunction of the neuroectoderm and cutaneous ectoderm) to the skin in its regression through the prenasal space, a dermal sinus tract is created, leaving a dimple at its orifice. The dermal sinus tract may extend superiorly along the path of the dural diverticulum for variable distance, and can communicate with the intracranial cavity. Clinical findings of the dermal sinus tract include the presence of a dimple or pit ( Fig. 9 ) over the nasal bridge near the osteocartilaginous junction (or anywhere from the glabella to the tip of the nose), hairs that emanate from the pit, and sebaceous discharge. Some of the tissue within the sinus dermal tract can create dermoid and/or epidermoid cysts, due to persistent ectodermal elements at sites of suture closure, brain diverticulation, and neural tube closure. These cysts can also be found along the following midline locations: the anterior fontanelle, glabella, nasion, vertex, subocciput, at the level of foramen cecum (adjacent to the crista galli), and adjacent to the anterior margin of the third ventricle.
MR imaging is the best choice to achieve the objective sought in studying these lesions, namely, evaluating the nature (imaging features of the tissue), locating site of origin, and determining the extension of the lesion.
Nasal gliomas are evident near the root of the nose, located paramedian to the bridge of the nose and external to the nasal cavity, while intranasal gliomas are inside the nasal cavity medial to the middle turbinate bone.
MR imaging is the modality of choice for the evaluation of these lesions, which are hypointense to isointense relative to gray matter on T1-weighted and T2 -weighted imaging, with a focal high signal on T2-weighted sequences within the lesion. This latter finding is due to gliosis, cysts, and myxoid degeneration. Even though the dysplastic tissue does not enhance, the surrounding compressed mucosa does. To demonstrate the intracranial stalk, a high-resolution surface coil MR imaging method must be applied.
Concerning the dermal sinus tract, not only the features of the sinus tract but also the associated dermoid and epidermoid cysts must be detected. The dermal sinus tract is seen as a hypointense line on T1-weighted images within the subcutaneous tissue of the midline nasal bridge, extending toward the prenasal space.
The epidermoid cysts are isointense to CSF on both T1-weighted (low signal) and T2-weighted (bright signal) images, and show restriction on isotropic diffusion-weighted images.
Because dermoid cysts have fat components, they demonstrate T1 shortening (bright signal) and variable hyperintensity on T2-weighted images, as well as variable signal intensity on diffusion-weighted images.
A bifid anterior crista galli and an enlarged foramen cecum, plus atypical signal intensity at the foramen cecum, indicate the presence of an intracranial epidermoid or dermoid cyst.
All of the aforementioned lesions can be surgically resected. The nasal dermoid and epidermoid cysts are operated on to avoid potential CNS infection if there is intracranial connection.
Anomalies of the corpus callosum
The primitive lamina terminalis is part of the rostral wall of the telencephalon, after closure of the anterior neuropore at about 25 days GA. During the seventh week GA, the dorsal end (near the paraphysis) of the lamina terminalis thickens and is called the lamina reuniens or commissural plate. The sulcus medianus telencephali medii is a groove developed in the lamina reuniens. The ventral surface of this groove is filled with cellular material from the developing subarachnoid space (meninx primitiva). A bridge or “sling” is formed across the sulcus medianus, probably of this material and of neurons from the developing hemispheres. The cells within this sling express surface molecules and secrete chemicals, which are required to guide the axons across the midline. During the 10th week GA, pioneer axons of different commissures cross the midline in the following order: first the anterior commissure (10th week), second the hippocampal commissure (11th week), and third the corpus callosum. The corpus callosum is sequentially formed, starting with the posterior genu, anterior body, posterior body and anterior genu, the splenium, and finally the rostrum. This knowledge is important in order to differentiate the callosal anomalies, as listed in Table 3 . Callosal anomalies are associated with anterior and hippocampal commissures anomalies, as mentioned in Table 4 . According to the injury during embryologic development, Utsunomiya and colleagues summarized callosal anomalies as follows: (a) total defect of corpus callosum, anterior commissure, and psalterium due to failure of lamina reuniens at 6 to 8 weeks GA; (b) total defect of corpus callosum due to failure of callosal precursor at 8 to 10 weeks GA; (c) partial defect of corpus callosum due to partial failure of callosal precursor at 10 to 15 weeks GA; (d) callosal hypoplasia due to failure to association fibers entering massa commissuralis at 11 to 20 weeks GA; (e) atrophy due to failure of commissural fibers in the corpus callosum at 20 weeks GA until after birth.
| Anomaly | Corpus Callosum | |
|---|---|---|
| Present | Absent or Small | |
| Agenesis: complete absence | None | All |
| Hypogenesis: incomplete formation | Posterior genu Anterior body | Anterior/inferior genu Posterior body Splenium, rostrum |
| Destructive | Splenium, rostrum | Genu, body |
| Dysgenesis: defective development (holoprosencephaly) | Splenium Genu, splenium Body, splenium | Genu, body, rostrum Body Genu |
| Corpus Callosum | Other Commissures |
|---|---|
| Agenesis | Anterior: small, absent |
| — | Hippocampal: small, absent, enlarged |
| Hypogenesis | Hippocampal: enlarged |
The development of the corpus callosum is not completed in utero. During the neonatal period and infancy, both postnatal loss and myelination of callosal axons occur. The etiology of corpus callosum dysgenesis is not completely known. Some implicated risk factors are listed in Box 7 .
- 1.
Irradiation
- 2.
Deficiencies in riboflavin, folic acid, niacin
- 3.
Maternal infection: rubella, congenital toxoplasmosis
- 4.
Fetal alcohol syndrome
- 5.
Maternal diabetes
Asymptomatic agenesis of the corpus callosum has been reported when found isolated; its incidence is very rare. The more frequent manifestations include seizures, macrocephaly, delayed development, mental retardation, and hypothalamic dysfunction. Dysgenesis of corpus callosum is commonly associated with many syndromes and other cerebral and cerebellar anomalies, such as other telencephalic commissure anomalies, Chiari II malformation, Dandy-Walker malformation, interhemispheric cysts, malformations of cortical development (neuronal migration and organization), cephaloceles, and midline facial anomalies.
MR imaging is an excellent modality for the assessment of the dysgenesis of the corpus callosum, because it displays not only the features of this anomaly but also other associated ones.
On axial images the following findings are evident: lateral convexity of the frontal horns, parallel lateral ventricles, colpocephaly (disproportionate dilatation of trigones and occipital horns of the lateral ventricles), keyhole dilatation of the temporal horns (deficient cingulate fasciculus), upward extension of the third ventricle into the interhemispheric fissure between lateral ventricles, communication of the third ventricle with the interhemispheric fissure anteriorly, interdigitation of gyri across the midline (due to partial absence of the falx in the interhemispheric fissure), and hypoplasia of the anterior commissure ( Figs. 10 and 11 ).
Sagittal images are the best sequence for the evaluation of the extent of the dysgenesis of the corpus callosum, which could be absent or incomplete ( Fig. 12 ).
On coronal images other features are well shown, such as persistent everted cingulated gyri, medial hemispheric sulci extending into the third ventricle, crescent-shaped lateral ventricles (due to impression on the medial walls of the ventricles by the medially positioned bundles of Probst), incomplete inversion of the hippocampus, and extension of the third ventricle into the interhemispheric fissure. Coronal images also demonstrate that the hippocampal commissure unites the fornices, not the cerebral hemispheres.
The longitudinal callosal bundles of Probst are formed when the axons cannot cross the midline, and are forced to lie laterally to the cingulate gyri and medially to the medial walls of the lateral ventricles, and parallel to the interhemispheric fissure, merging (by its inferomedial borders) with rudimentary fornices. These bundles are brighter on T1 and darker on T2 images than are other white matter fibers. MR imaging findings of corpus callosal dysgenesis are mentioned in Box 8 .
- 1.
White matter abnormalities
- a.
Probst bundles
- b.
Anterior commissure hypoplasia
- a.
- 2.
Gyral and sulcal abnormalities
- a.
Persistent eversion of cingulated gyrus
- b.
Absent cingulated sulcus
- c.
Hippocampal hypogenesis
- a.
- 3.
Ventricular abnormalities and cysts
- a.
Widened third ventricle, superiorly extended into interhemispheric cyst
- b.
Colpocephaly, crescentic shape of lateral ventricles on coronal images, parallel or lateral convexity of lateral ventricles on axial images
- a.
The lack of formation or destruction of the callosal axons correlate with markedly reduced volume of cerebral white matter. If the callosal anomaly is secondary to an improper guidance of axons across the midline, the volume of the cerebral cortex and the appearance of white matter are normal.
Agenesis of corpus callosum presenting interhemispheric cyst (IHC) is classified by Barkovich into two major groups: Type 1, diverticulum of the ventricular system that communicates with the ventricles; and Type 2, multiple cysts, which do not communicate with the ventricles. Type 1 and Type 2 are divided into subgroups depending on the clinical manifestations and associated anomalies. Type 1 IHC is isointense to CSF. Type 2 IHC is slightly hyperintense to CSF on T1-weighted images and isointense to hyperintense on T2-weighted images. Subcortical heterotopia is seen in Type 2.
Intracranial lipomas
Intracranial lipomas are congenital malformations composed of abnormal collections of fat. Lipomas probably derive from abnormal differentiation of the meninx primitiva, which normally differentiates into the leptomeninges and the subarachnoid space.
The meninx primitiva originates from the mesoderm of the neural crest cells. First, the developing embryo is involved by the meninx primitiva, then (in an established sequence) the inner meninx resorbs, leaving the subarachnoid space. There is an inverse correlation between the pattern of cavitation and dissolution of the meninx with the allotment of intracranial lipomas: the last zone to resorb is the dorsum of the lamina terminalis, and lipomas are more frequent (40%–50%) at the pericallosa area. Other sites for lipomas are the quadrigeminal plate/superior cerebellar cisterns (25%), the suprasellar/interpeduncular cisterns (10%–20%), the cerebellopontine angle cisterns (7%), and the Sylvian cisterns (5%). Blood vessels and cranial nerves course through these lipomas (within the subarachnoid space). Intracranial lipomas are usually asymptomatic and are considered incidental findings, even though some patients present with seizures or cranial neuropathy secondary to involvement of the adjacent neural tissue. Lipomas are habitually juxtaposed to malformed brain, such as callosal lipomas ( Fig. 13 ) near callosal hypogenesis, frontal interhemispheric lipomas associated with midline facial clefts, and nasofrontal or nasoethmoidal cephaloceles; even extension of the lipoma into the cephalocele is frequently seen. The term midline craniofacial dysraphism refers to a midline developmental disorder associated with a lipoma of the interhemispheric fissure. Intracranial lipomas are malformations (not tumors) and they increase in bulk only with somatic growth.
Related posts:
 Demystifying Congenital Malformations for Imaging Specialists
Demystifying Congenital Malformations for Imaging Specialists
 Development and Dysgenesis of the Cerebral Cortex: Malformations of Cortical Development
Development and Dysgenesis of the Cerebral Cortex: Malformations of Cortical Development
 Imaging of Congenital Spine and Spinal Cord Malformations
Imaging of Congenital Spine and Spinal Cord Malformations
 Neurosurgical Management of Congenital Malformations and Inherited Disease of the Spine
Neurosurgical Management of Congenital Malformations and Inherited Disease of the Spine
 Congenital Malformations of the Temporal Bone
Congenital Malformations of the Temporal Bone
 Hemangiomas and Vascular Malformations of the Head and Neck: A Simplified Approach
Hemangiomas and Vascular Malformations of the Head and Neck: A Simplified Approach
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


