Abstract
Miller-Dieker syndrome, or 17p13.3 deletion syndrome, is a rare, contiguous gene deletion syndrome characterized by type 1 lissencephaly, facial dysmorphism, seizures, and severe mental retardation. Impaired neuronal migration between 6 and 15 weeks’ gestation is the underlying mechanism for lissencephaly, leading to the absence of sulci and gyri on the surface of the brain as well as abnormal cortical thickness. Heterozygous deletions in the region of chromosome 17p13.3 result in the severe phenotype of Miller-Dieker syndrome. Suspicious sonographic findings include absent parietooccipital and calcarine fissures as well as a “smooth, shallow sylvian fissure.” Associated findings include ventriculomegaly, agenesis of the corpus callosum, fetal growth restriction, and polyhydramnios. Fetal MRI can be a useful adjunct for prenatal diagnosis, with the characteristic finding of a smooth brain surface with an abnormally shallow sylvian fissure. Miller-Dieker syndrome is incurable, with no known prenatal treatment options. Postnatal treatment consists largely of antiepileptic therapy for seizure control and supportive care. Suspicion for Miller-Dieker syndrome should be raised in patients with a family history and/or imaging findings relating to lissencephaly. Parental chromosomal analysis is indicated at the time of diagnosis, as 20% of deletions are inherited from a parent with a balanced chromosomal rearrangement.
Keywords
Miller-Dieker syndrome, lissencephaly, 17p13.3 deletion syndrome, LIS1 gene
Introduction
Miller-Dieker syndrome (MDS) is a rare, contiguous gene deletion syndrome characterized by type I lissencephaly, facial dysmorphism, seizures, and severe mental retardation. Other associated defects including cardiac malformations, neural tube defects, omphalocele, gastrointestinal anomalies, genitourinary anomalies, and intrauterine growth restriction have also been described in relationship to this syndrome. MDS is caused by a deletion at the chromosome 17p13.3 locus and is thought to represent the severe end of the spectrum of classical lissencephaly. The prognosis for affected patients is poor, with mortality typically occurring within the first 1 to 2 years of life.
Disorder
Definition
Lissencephaly is a defect in neuronal migration characterized by a smooth cerebral surface, either with agyria (absent gyri) or pachygyria (abnormally broad brain folds), a thickened cerebral cortex, and an increased ratio of gray to white matter. Mutation in the causative LIS1 gene leads to a spectrum of disorders encompassing isolated lissencephaly, subcortical band heterotopia, and MDS. Patients with MDS tend to have larger deletions, resulting in more severe lissencephaly in combination with craniofacial dysmorphisms, as well as other associated anomalies.
Prevalence and Epidemiology
Due to the rarity of disease, the prevalence of MDS is unknown. At least 29 cases of MDS with the 17p13.3 deletion and prenatal findings have been reported in the literature. Classical lissencephaly is reported to occur in 11.7–40 : 1 million births.
Etiology, Pathophysiology, and Embryology
Impaired neuronal migration between 6 and 15 weeks’ gestation is the underlying etiologic mechanism for lissencephaly. Postmitotic neurons are unable to migrate to their final destination and therefore do not correctly populate the cortical plate of the cerebral cortex. This leads to the absence of sulci and gyri on the surface of the brain, as well as abnormal cortical thickness.
Heterozygous deletions in the region of chromosome 17p13.3 result in the MDS phenotype. Located within this region is the responsible lissencephaly gene (LIS1 or PAFAH1B1), which has been highly conserved throughout evolution. This gene encodes the β subunit of the platelet-activating factor acetylhydrolase brain isoform, which inactivates platelet-activating factor, a neuroregulatory molecule. While deletions in LIS1 are observed in both isolated lissencephaly and MDS, it is thought that the more severe phenotype observed in MDS is caused by a combined genetic effect from deletions in both LIS1 as well as the YWHAE gene, located about 40 kb telomeric to LIS1 within a region known as the “MDS telomeric critical region.” Loss of the YWHAE gene, not including LIS1 , results in facial dysmorphism, intrauterine growth restriction (IUGR), and neuroradiologic changes. Large deletions that extend farther toward the 17p telomere, including the YWHAE gene, result in the more severe phenotype as observed in MDS. Approximately 80% of patients with MDS have a de novo deletion in the 17p13.3 region, and the remaining 20% are thought to inherit the deletion from a parent who carries a balanced chromosomal translocation.
Manifestations of Disease
Clinical Presentation
The clinical features of MDS include profound neurodevelopmental delay, seizure activity, failure to thrive, and feeding difficulties. Infants are typically hypotonic at birth and subsequently develop spasticity. The classic facial dysmorphisms include a prominent forehead, low-set ears, widely spaced eyes, a thickened upper lip with a thin vermillion border, an elongated philtrum, a short, upturned nose, bitemporal narrowing, and micrognathia. Other variable findings include microcephaly, clubfoot, polydactyly, cleft lip/palate, omphalocele, duodenal atresia, hepatosplenomegaly, sacral dimples, joint contractures, hypoplastic genitalia in affected males, and renal and cardiac malformations.
Imaging Technique and Findings
Ultrasound.
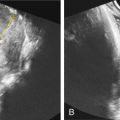
The sonographic evaluation for lissencephaly at the time of the midtrimester anatomic survey poses a diagnostic challenge given that brain development and maturation, including formation of normal cerebral gyration, continues throughout gestation. Suspicious sonographic findings include an absent parietooccipital and calcarine fissure as well as a smooth, shallow sylvian fissure, which can be detected in the standard plane used to measure biparietal diameter ( Fig. 157.1 ). These findings have been reported on prenatal ultrasound (US) as early as 23 weeks’ gestation. It is important to note that prior to 20 weeks’ gestation, it is normal for the sylvian fissure to appear as a smooth, shallow depression. Other central nervous system (CNS) findings such as ventriculomegaly and agenesis of the corpus callosum (ACC) can be markers for lissencephaly. IUGR and polyhydramnios (secondary to impaired fetal swallowing) are common findings. In the 29 documented cases of MDS with prenatal findings, polyhydramnios, IUGR, and ventriculomegaly were the most common associated sonographic findings, each seen in approximately 2 out of 3 of cases. With the exception of micrognathia, the characteristic facial dysmorphisms are difficult to detect with sonographic imaging.

Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


