- •
Nuclear cardiology currently evolves from assessing physiologic function and perfusion toward an interrogation of molecular pathways and pathophysiologic alterations at a (sub)cellular level.
- •
Given a biphasic inflammatory response post-MI (acute inflammation followed by a reparative phase), targeted molecular imaging could be helpful in identifying the right patient for the right treatment at the right time.
- •
These considerations are further fueled by multiple cardiac-paired diagnostic agents and matched therapy, eventually leading to systems-based, image-guided therapy.
- •
In this regard, novel molecular imaging–guided concepts for cardiac repair have emerged in recent years, which target chemokine receptors, SSTRs, MMPs, TSPOs, FAPs, and angiogenesis markers.
- •
Future developments in the field include enhanced visualization of therapeutic drug targets and imaging-guided drug development.
Introduction
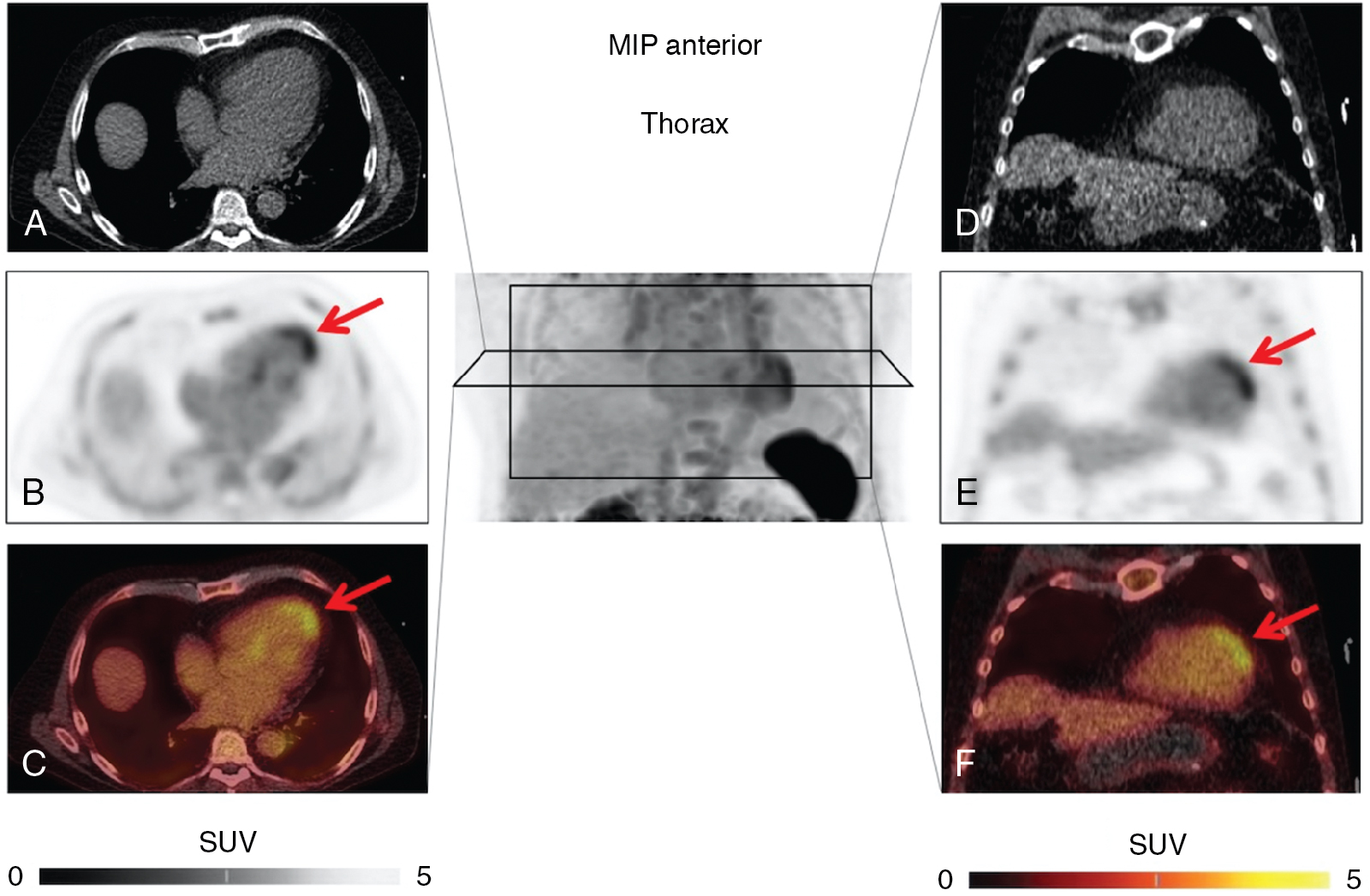
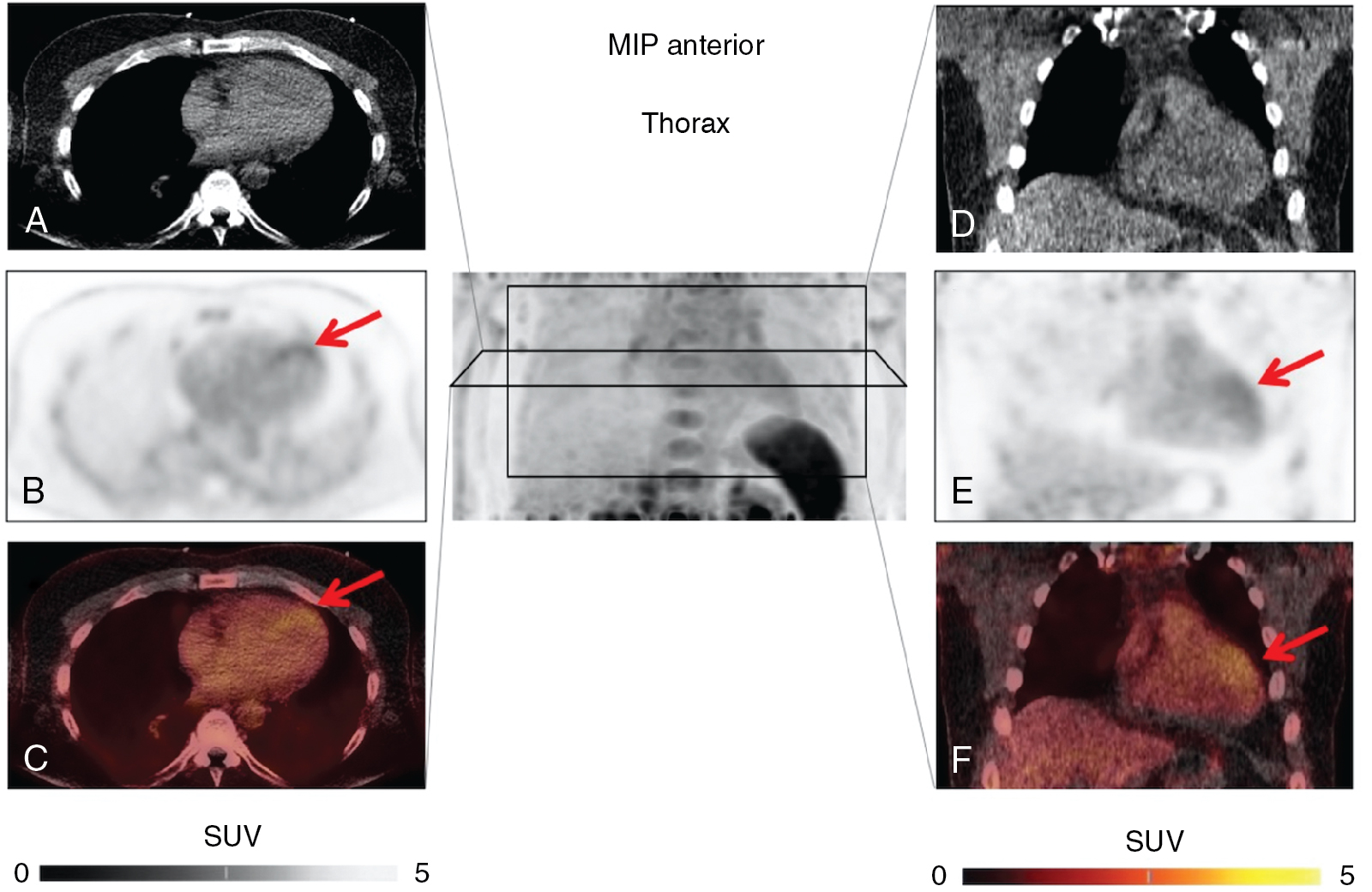
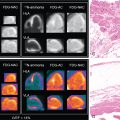
Current treatment strategies, including early revascularization, have significantly improved patient survival after acute myocardial infarction (AMI). Despite optimal medical therapies, however, survivors are at an increased risk for post–myocardial infarction (MI) heart failure (HF). Conventional management of patients with HF post-MI include a broad range of systemic therapies designed to attenuate the risk for adverse left ventricular (LV) remodeling. This one-size-fits-all approach for treatment in HF after MI could benefit from a more personalized, targeted approach. These considerations are further fueled by emerging targets involved in myocardial repair after MI, including inflammatory/immune cells, fibroblasts, and cardiac progenitor cells. Some novel targeted therapies, however, which demonstrated promising results in early testing and a preclinical environment, failed to translate into meaningful clinical benefit, , and others demonstrated promising results in lowering recurrent cardiovascular events. Such striking patient-to-patient heterogeneity emphasizes the opportunity for tailoring novel targeted drugs to the individual patient’s characteristics. In this regard, the expanded use of “liquid biopsies” is helping to identify patient-specific pathophysiologic makeup by investigating components of the myocardial injury/repair process that are shed from the heart into the circulatory system. Although liquid biopsies hold the promise to overcome certain drawbacks compared with conventional tissue-based biopsies, such approaches neglect intrinsic heterogeneity or tissue-specific information or variations. Thus noninvasive molecular imaging, which identifies even modest alterations at a (sub)cellular level may meet the increasing demands associated with precision medicine by providing critical insights into the underlying pathophysiologic process that affects the development of HF after AMI. Thus, applying molecular imaging combined with specific radiotracers may open avenues for tailoring the initiation of novel drugs for myocardial repair and, ideally, allow for optimal-effective dose determination strategies after the acute event. For instance, the peptidic macrocycle C-X-C motif chemokine receptor 4 (CXCR4) antagonist POL5551 and its clinical stage analog POL6326 modulated the immune response to injury, enhanced tissue repair, and led to an improvement of mechanical function after AMI in both mice and pigs. In addition, there is evidence that another CXCR4 antagonist, AMD3100, prolongs bone marrow progenitor mobilization, improving functional recovery in a murine model of transient coronary occlusion. These encouraging preclinical results have paved the way for initiation of clinical trials investigating the effects of CXCR4 antagonists as stem cell mobilizing agents in patients after AMI. Interestingly, the novel CXCR4-targeted positron emission tomography (PET) probe, 68 gallium (Ga)-Pentixafor, could potentially be used as a diagnostic counterpart for CXCR4-targeted therapy and may identify the patients who will be the most likely to respond to this therapy. In this regard, distinct patient heterogeneity of the Pentixafor signal has been reported in patients after AMI, ranging from intense CXCR4 upregulation in the infarct territory ( Fig. 26.1 ) to no discernible radiotracer accumulation after MI ( Fig. 26.2 ). The large heterogeneity in signal strength in the area at risk may be caused by multiple factors, including the timing of imaging after AMI, the response to concomitant medications or differences in CXCR4 activation, and the selective recruitment of tissue-specific leukocytes. Nonetheless, the observed heterogeneous uptake pattern in the infarct territory may provide a guide for the timing of initiation of small molecular inhibitors of CXCR4 during the peak of cardiac uptake.
Molecular repair mechanisms after acute myocardial infarction: Inflammation, fibrosis, and angiogenesis
Established molecular imaging-guided concepts
To date, 18 F-fluorodeoxyglucose ( 18 F-FDG) is the only radiotracer approved by the U.S. Food and Drug Administration for the evaluation of myocardial viability in ischemic cardiomyopathy. As discussed in Chapters 19 and 20 , the underlying concept is based on the recognition that ischemic cardiomyopathy can result from myocardial scar (i.e., irreversible injury of cardiomyocytes) or hibernating myocardium (i.e., temporary loss of contraction ability within an area of viable tissue). As such, LV dysfunction may be reversible from hibernation by revascularization procedures, underpinning the importance of reliably segregating between ischemically affected but still viable tissue and scar formation. 18 F-FDG PET-assisted management in patients with coronary artery disease (CAD) serves as a gatekeeper to the catheterization laboratory and has substantially improved outcome with revascularization. Nonetheless, the current approach of considering flow-metabolism mismatch as ischemic but still viable has inherent limitations. , First, cardiac glucose metabolism may be altered by multiple patient-specific factors, such as dietary state, blood glucose level, cardiac workload, or sympathetic tone. Second, a considerable number of patients referred to PET centers to conduct myocardial viability assessments with 18 F-FDG are afflicted by insulin resistance from diabetes and/or HF and even after oral glucose loading, the amount of endogenous insulin may be insufficient to achieve maximal stimulation of 18 F-FDG uptake. A hyperinsulinemic euglycemic clamp may overcome this issue by switching insulin-sensitive tissues to glucose use, but the challenging and time-consuming nature of this technique limits its more widespread adoption in the clinic. Finally, variable uptake of 18 F-FDG by inflammatory cells or activated fibroblasts in the ischemically injured region may also confound the interpretation of FDG retention, especially early after AMI. This is supported by the fact that the magnitude of increased glucose utilization in the culprit territory 7 days after MI is associated with activation of lymphoid tissue in spleen and bone marrow. Consequently, there is a need for more specific approaches targeting inflammation, especially as novel and highly innovative therapies for myocardial repair, such as specific anti-inflammatory drugs or even T cell–based immunotherapies, are being developed. , This opens new opportunities for targeted molecular imaging approaches to evaluate the timing and efficacy of novel reparative interventions ( Fig. 26.3 ).
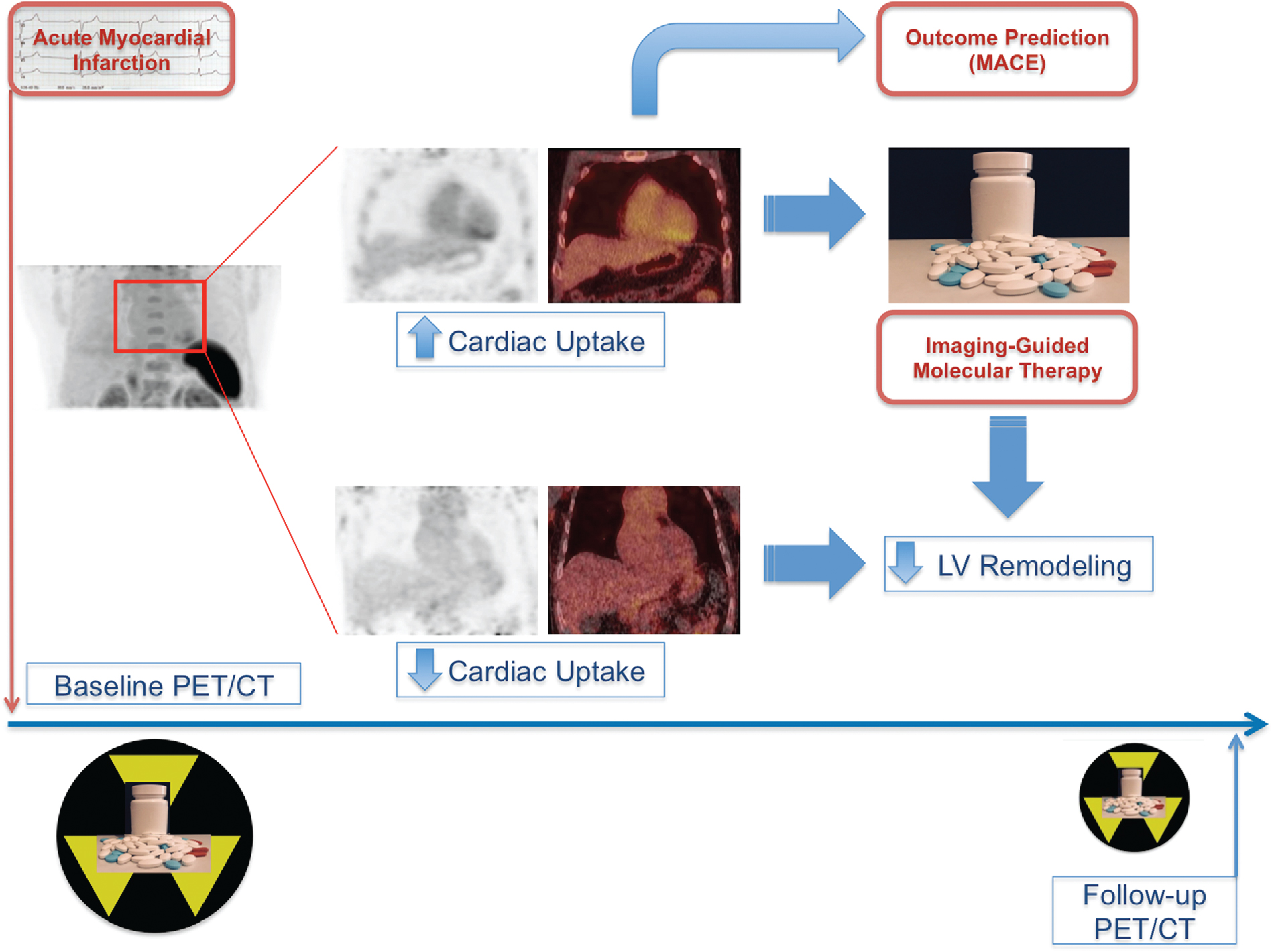
Molecular reparative interventions after acute myocardial infarction
Myocardial revascularization of the culprit vessel is an established therapy for patient management early after AMI. Despite successful revascularization, more than one-third of the patients will develop late-onset HF, mainly caused by progressive LV remodeling. , Stem cell therapy for HF complications has yielded mixed results with modest effect on clinical outcomes, including mortality or rehospitalization. , Given the rather limited beneficial effect of autologous stem/progenitor cells for promoting cardiac regeneration, cardiac cell therapy has not entered clinical use. Interestingly, recent studies demonstrated that the sustained improvement of cardiac function after cell therapy is primarily driven by an acute inflammatory-based wound-healing response. As such, acute inflammation may be an important but yet underrecognized aspect of tissue healing after ischemic injury. Nevertheless, chronically persistent inflammation is thought to contribute to adverse remodeling. Early after AMI, migration of neutrophils and inflammatory monocytes into the ischemic area facilitate phagocytosis of cellular debris resulting from ischemic injury. This is followed by a reparative phase with resolution of inflammation, fibroblast proliferation, and scar formation. Prolongation or insufficient suppression of this inflammatory phase can lead to sustained tissue damage and inadequate healing, thereby promoting infarct expansion, chamber dilatation, and adverse remodeling. Immune-modulating therapeutic strategies that favorably influence wound healing and repair after AMI are being investigated. , In patients with stable CAD without HF, the results of studies targeting a reduction in inflammation have been mixed. For instance, in the Cardiovascular Inflammation Reduction Trial (CIRT), the broad-spectrum antiinflammatory agent methotrexate did not improve clinical outcomes among stable CAD patients with either diabetes or metabolic syndrome. It is worth noting that an elevated C-reactive protein (CRP) level was not required for recruitment in CIRT and that the median CRP value in the cohort was mildly elevated (1.6 mg/L) and did not change after treatment with low-dose methotrexate. In contrast, the Canakinumab Anti-Inflammatory Thrombosis Outcome Study (CANTOS) trial required an elevated CRP level (median value: 4.3 mg/L). It showed that highly specific interleukin (IL)-1 beta inhibition was effective at reducing inflammation, which resulted in a significant reduction in recurrent cardiovascular events in stable CAD patients with previous MI. The apparently divergent observations highlight the importance of considering the diversity of inflammatory pathways in atherosclerosis and the potential means to their modulation. It also remains unclear whether targeted antiinflammatory therapy can be beneficial early after AMI to support wound healing and clinical outcomes. Given the biphasic inflammatory response after MI, targeted molecular imaging could be helpful in identifying the right patient population and understanding the efficacy of such therapy in clinical trials.
Novel molecular imaging–guided concepts
Several novel molecular imaging–guided concepts for cardiac repair have emerged in recent years, including therapeutic targets of inflammation, angiogenesis, and fibrosis.
Chemokine receptors
CXCR4/stromal cell–derived factor (SDF)-1 alpha tightly orchestrates the recruitment and homing of stem and progenitor cells to injured myocardium after MI. In recent years, a growing body of evidence has reported on the use of the radiolabeled CXCR4 ligand 68 Ga-Pentixafor, which allows for high-contrast visualization of the presence of the receptor in vivo. In mice, Thackeray et al. demonstrated that 68 Ga-Pentixafor described the dynamic changes in CXCR4 upregulation, which peaked at 3 days after MI and declined at 7 days. The early CXCR4 signal was proportional to leukocyte content in the LV myocardium. Treatment with the angiotensin-converting enzyme (ACE) inhibitor enalapril attenuated the signal, whereas flow cytometry demonstrated a similar temporal reduction in ventricular leukocyte content. Importantly, CXCR4 upregulation at day 3 independently predicted acute ventricular rupture and subsequent adverse LV remodeling over 6 weeks. CXCR4 blockade improved late ejection fraction (EF) when applied during peak upregulation but not off-peak, highlighting the importance of understanding the dynamic changes in CXCR4 upregulation to define the optimal timing of therapy. Further supporting the concept of image-based guidance for timing initiation of CXCR4 blockade, Jujo et al. demonstrated that a single-dose injection of the small molecule CXCR4 antagonist AMD3100 administered after the onset of MI in mice improved cardiac function and survival, whereas continuous infusion of AMD3100 worsened outcomes. The use of 68 Ga-Pentixafor in patients shortly after AMI also identified heterogeneity in CXCR4 upregulation (see Figs. 26.1 and 26.2 ), which further supports the notion of a molecular imaging–guided treatment strategy for identifying the ideal time for on-peak treatment to maximize efficacy. , The recent introduction of the theranostic equivalent of 68 Ga-Pentixafor ( 177 Lu-/ 90 Y-labeled Pentixather) to treat hematologic malignancies demonstrated an associated antiinflammatory effect on atherosclerotic plaques in the vessel wall. Thus, these feasibility studies may serve as a stimulus for further evaluation of radionuclide-based approaches for directing and lowering the degree of inflammation in high-risk atherosclerosis or after MI, preferably by implementing a dosimetry-based concept to maximize treatment efficacy by minimizing potential harm.
Furthermore, alternative chemokine receptors are currently emerging as potential targets for noninvasive molecular imaging after MI. For instance, 68 Ga-DOTA–labeled extracellular loop 1 inverso ( 68 Ga-DOTA-ECL1i) allows for imaging of proinflammatory CCR2+ monocytes/macrophages in the ischemically injured heart via binding of the ECL1i peptide to the extracellular domain of CCR2. Compared with the rather ubiquitous expression of CXCR4, CCR2 is mainly overexpressed on a subset of tissue-resident cardiac macrophages contributing to HF progression, thereby rendering CCR2 as an attractive and more specific imaging target after myocardial injury. Indeed, autoradiography studies of human heart sections of subjects afflicted with HF confirmed the high specificity of CCR2-targeted imaging. Thus 68 Ga-DOTA-ECL1i may also allow for the profiling of the extent of inflammatory overshooting and the identification of the therapeutic window for specific interventions, in particular, where multiple clinical trials testing broad antiinflammatory and immunomodulatory agents have failed in patients with MI.
Somatostatin receptors
Primarily used for neuroendocrine tumors of the gastroenteropancreatic region, membrane-bound somatostatin receptors (SSTRs) on the tumor cell surface can be targeted by molecular imaging using 68 Ga-1,4,7,10-tetraazacyclododecane-N,N’, N”,N”’-tetraacetic acid-d-Phe(1)-Tyr(3)-octreotide/-octreotate ( 68 Ga-DOTATOC/-TATE). High levels of the SSTR subtype 2 occur predominantly in proinflammatory M1 macrophages. Preclinical studies, however, yielded mixed results in macrophage-rich aortic atherosclerotic plaques in mice. The high blood pool activity is most likely caused by slow blood clearance, suggesting that SSTR-targeted PET may be a less attractive alternative for identifying macrophage-rich sites in cardiovascular disease. These observations were further confirmed in a mouse model of MI that demonstrated rapid blood clearance and extremely low standardized uptake values in the area of myocardial injury. Despite modest results in animals, application of SSTR-PET/computed tomography (CT) in patients afflicted with either myocarditis or subacute MI showed a close spatial relation of SSTR-targeted macrophages and structural changes assessed by cardiac magnetic resonance (CMR) imaging. As discussed in Chapter 12 , 68 Ga-DOTATATE PET/CT offers excellent macrophage specificity with relatively high SSTR2 expression in culprit carotid lesions and outperformed 18 F-FDG for the discrimination of high-risk versus low-risk coronary lesions. Apart from stable atherosclerosis, the same research group demonstrated that SSTR-directed imaging also identifies active inflammation in infarcted myocardium, which may further expand the clinical spectrum of SSTR PET to patients afflicted with recent AMI.
Matrix metalloproteinases
Increased matrix metalloproteinase (MMP) activity has been associated with numerous cardiovascular diseases, including MI, atherosclerosis, and dilated cardiomyopathy. In animal models, administration of MMP inhibitors has had beneficial effects on pump dysfunction, healing after MI, and ventricular remodeling by regulating extracellular matrix turnover and inflammatory signaling. Nevertheless, broad-spectrum or selective inhibitors of MMP used in post-MI clinical trials were inconclusive and failed to reduce LV remodeling or improve clinical outcomes. In a pig model of MI, the 99m technetium (Tc)-labeled MMP–targeted radiotracer RP805 showed increased retention early after MI, along with a high regional concordance with ex vivo MMP2 activity. The recently developed pan-MMP inhibitor–based single photon emission computed tomography (SPECT) tracer 99m Tc-RYM1 also enabled specific visualization of MMP inflammatory activity in carotid aneurysm. RP782, an 111 indium (In)-labeled SPECT compound, was used to detect injury-induced MMP activation in murine carotid arteries; increased radiotracer accumulation in the injured vessel wall was confirmed by autoradiography. Apart from the potential use of MMP-directed imaging in atherosclerosis, 111 In-RP782 holds potential for visualizing MMP-related post-MI remodeling early after the acute event. Given the intrinsic advantages of PET technology, including higher spatiotemporal resolution, the MMP-targeted PET probe 18 F-IPFP was recently introduced for application in pulmonary imaging and could also potentially be applied to MI or subsequent HF. Nevertheless, feasibility of MMP imaging for characterization of the inflammatory burden after MI or subsequent ventricular remodeling requires further studies. Well-designed and executed clinical trials may reveal the true potential of the derived imaging signal for tracking effects of therapeutic interventions (e.g., application of tissue inhibitors of metalloproteinases [TIMP analogs]).
Mitochondrial translocator protein
The 18 kDa mitochondrial translocator protein (TSPO) plays a crucial role in the regulation of innate immunity and is overexpressed on the mitochondrial membrane, especially in activated macrophages. TSPO-targeted radiotracers, such as 11 C-PK11195, can image inflammation in carotid atherosclerotic plaques. The TSPO ligand N,N -diethyl-2-[4-(2-fluoroethoxy)phenyl]-5,7-dimethylpyrazolo[1,5- a ]pyrimidine-3-acetamide ( 18 F-DPA-714) enabled visualization of the macrophage-mediated immune rejection of transplant-induced pluripotent stem cell–derived cardiomyocytes of allogeneic origin on the LV in mice. CD68+ macrophages and CD3+ T-lymphocytes were accumulated in the same regions of the grafted tissues and correlated with the PET-derived signal strength. TSPO-targeted whole-body molecular imaging may also provide further insights into the interplay between interconnected pathophysiologic pathways of the inflamed myocardium after MI and disparate organs. , Thackeray et al. reported on concomitant upregulation of the macrophage- and microglia-expressed TSPO in heart and brain after AMI using the novel PET imaging agent 18 F-flutriciclamide ( 18 F-GE180). The acute TSPO signal in the infarct territory independently predicted contractile dysfunction at 8 weeks after coronary ligation and after a transient decline, remote myocardial and brain TSPO was reelevated in chronic HF. Nonetheless, TSPO PET for imaging of AMI is challenging, and second-generation radiotracers may overcome current drawbacks, such as high background signal, because of high mitochondrial content in the heart, nonspecific binding, and genetic variability, mainly caused by TSPO gene polymorphisms. In addition, TPSO PET may also guide novel therapies, such as on-peak treatment with the cardioprotective TSPO ligand 4´-chlorodiazepam.
Fibroblast activation protein
Myocardial fibroblasts play a pivotal role in mediating the reparative response after MI. Activated fibroblasts migrate into the infarct territory and maintain the extracellular matrix in the scar, thereby contributing to an effective repair of the cardiac interstitium by promoting fibrosis formation for stabilization of the LV wall. Nevertheless, persistence of activated fibroblasts and deposition at sites remote from the infarct area can lead to worsening myocardial stiffness and progression to HF. As a homodimeric membrane-bound serine protease, fibroblast activation protein (FAP) is overexpressed by activated fibroblasts and was identified in human hearts after MI. Recently, radiolabeled FAP inhibitors (FAPIs) have been introduced for imaging of various cancer entities and, relative to 18 F-FDG, these novel FAPI imaging agents have inherent advantages, including the ability to acquire images right after injection and the lack of need for dieting or fasting preparation before the scan. As such, imaging of activated fibroblasts using the 68 Ga-labeled FAP inhibitor ( 68 Ga-FAPI-04) in a preclinical model of MI revealed intense FAP expression in the injured myocardium; immunofluorescence staining confirmed the presence of FAP-positive myofibroblasts in the border zone. In patients with advanced HF, however, myofibroblasts retained the capacity to return to less activated states but also exhibited variable degrees of dedifferentiation. This heterogenous population of fibroblastic cells in the LV of end-stage HF patients further emphasizes the need for an imaging-guided therapeutic approach to identify the optimal time window to enhance potential responsiveness of antifibrotic therapy. For instance, differentiation of fibroblasts is dependent on transforming growth factor beta, but inhibition after ischemic injury increased mortality and worsened outcome. In a dedicated murine model, considerable cardiac fibrosis followed by subsequent dysfunction was induced and the beneficial effects of redirected T-cell immunotherapy to specifically target cardiac fibrosis was evaluated. Cardiac fibroblasts expressing a xenogeneic antigen were not only effectively targeted but also ablated by adoptive transfer of antigen-specific CD8+ T cells. Such an adoptive transfer of T cells that express a chimeric antigen receptor against FAP significantly reduced cardiac fibrosis and improved outcome. Of note, this highly specific antifibrotic treatment approach could be effectively targeted by radiolabeled FAPIs, which may further increase therapeutic efficacy.
Angiogenesis marker
α v β 3 integrin is a transmembrane cell surface receptor critically involved in migration, proliferation, and interaction with the extracellular matrix. As shown by inhibition of blood vessel formation using antibodies, α v β 3 integrin is barely detectable in endothelial cells at quiescent state, but it is overexpressed in vascular endothelial cells, which makes this molecule an attractive target for monitoring angiogenesis in response to hypoxia. In addition, α v β 3 integrin is expressed by both activated cardiac fibroblasts and macrophages, which further emphasizes its preeminent role in coordinating repair after AMI. The 111 In-labeled radiotracer targeted at the α v β 3 integrin, RP748, effectively localized hypoxia-induced α v β 3 activation in the heart early after AMI, which emphasizes its potential to identify treatment responders for angiogenic-targeted therapies. Moreover, α v β 3 integrin contains a binding site for an Aginylglycylaspartic acid (RGD) subunit (the arginine-glycine-aspartate motif). Thus, PET using the 18 F-labeled glycosylated α v β 3 integrin antagonist Galacto-RGD may even overcome the limitations associated with SPECT. In a rat model of MI, 18 F-Galacto-RGD demonstrated increased uptake between 1 and 3 weeks, with sustained signal in the infarct zone 6 months after reperfusion, indicating a delayed but prolonged tissue response after ischemia reperfusion. Moreover, blocking studies confirmed tracer specificity for α v β 3 integrin expression and immunohistochemistry also revealed a close colocalization of the tracer signal with newly formed vessels. In the absence of LV remodeling, high levels of 18 F-Galacto-RGD were reported, suggesting that this PET compound can be used to monitor myocardial repair after AMI and provide prognostic information. , The increased uptake of the second-generation α v β 3 integrin-selective radiotracer 18 F-Fluciclatide in segments displaying functional recovery by cardiac MRI further supports its potential to predict adverse remodeling. Because α v β 3 integrin expression can be quantified in the infarct territory, 18 F-Fluciclatide may hold the potential to guide toward specific therapies with vascular growth factors, cell-based therapies, or other proangiogenic drugs.
Angiotensin-converting enzyme and angiotensin II receptor type 1 ligands
Multiple randomized controlled trials clearly established the beneficial effects of ACE inhibitors in HF with reduced ejection fraction; its use with other renin-angiotensin system (RAS) inhibitors is standard therapy in HF. The 11 C-labeled angiotensin II receptor type 1 (AT1R) ligand 2-butyl-5-methoxymethyl-6-(1-oxopyridin-2-yl)-3-[[2-(1H-tetrazol-5-yl)biphenyl-4-yl]methyl]-3H-imidazo[4,5b]pyridine ( 11 C-KR31173) is a methoxy analog of the nonpeptide AT1-selective antagonist SK108 and demonstrated peak myocardial uptake 1 to 3 weeks after MI in mice. Specificity of the radiotracer was proven by oral administration of the clinically established AT1R blocker valsartan. First-in-man application demonstrated a high safety profile, along with specific myocardial 11 C-KR31173 retention, which lay the groundwork for future studies monitoring the efficacy of anti-RAS directed therapy noninvasively. These considerations are further fueled by recent major clinical trials showing the high efficacy of sartans in combination therapies. For instance, a combined treatment of valsartan together with the neprilysin inhibitor sacubitril was superior to enalapril in reducing the risk of death and HF hospitalization. Nevertheless, radionuclide imaging with 11 C-labeled agents has several drawbacks, mainly because of its short half-life of 20 minutes. Thus a great deal of progress has been made by the introduction of novel 18 F-based radiotracers, which overcome some of these obstacles associated with short-lived PET agents. As such, the novel 18 F-labeled PET radiotracer FV45, which is based on a derivation from the clinically used drug valsartan with an almost identically chemical structure, has been recently introduced. Interestingly, this agent also demonstrated selective blockade of renal radiotracer accumulation by valsartan pretreatment, which renders 18 F-FV45 as an attractive compound for assessing renal diseases.
Table 26.1 provides an overview of currently established molecular imaging agents and their matched therapeutic counterparts, along with their current status for imaging and therapy. Fig. 26.4 summarizes relevant molecular targets after MI, which can be imaged by the referring PET/SPECT compounds.
Related posts:
 Radiopharmaceuticals for clinical SPECT and PET and imaging protocols
Radiopharmaceuticals for clinical SPECT and PET and imaging protocols
 Imaging in patients with acute chest pain in the emergency department
Imaging in patients with acute chest pain in the emergency department
 Key concepts in risk stratification and cost-effectiveness using nuclear scintigraphy in stable coronary artery disease
Key concepts in risk stratification and cost-effectiveness using nuclear scintigraphy in stable coronary artery disease
 Patient with known or suspected cardiac sarcoidosis
Patient with known or suspected cardiac sarcoidosis
 Artificial intelligence in nuclear cardiology
Artificial intelligence in nuclear cardiology
 Large-vessel vasculitis
Large-vessel vasculitis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


