Infections of the brain in the postnatal period differ from those in older children as a result of a combination of distinct epidemiologic factors in general, and immaturity of neonatal brain and immunologic host response in particular. It has been recognized that clinical and neurologic signs are often nonspecific, sometimes scarce, and seldom correlate with the extent of neuroimaging findings, thus warranting an early MR imaging examination in the course of the disease, enabling rapid therapy institution and better clinical outcome. This article reviews most of postnatal pathogen agents involved in neonatal brain infections, related physiopathology, and neuroimaging findings.
Infection of the central nervous system (CNS) in neonates may be caused by a variety of bacteria, viruses, fungi, or parasites. The late sequels of CNS insult vary with the amount and virulence of the pathogen agent, the timing of infection in relation to the degree of cerebral maturation, the amount of immunologic protection transmitted from the mother, and the ability of the neonate’s immune and inflammatory system to respond to the infection. Neuroimaging patterns not only reflect changes related to selective tissue injury as in many viral infections, but also result from nonspecific changes caused by a more generalized reaction of the host as seen mainly in bacterial infections. Furthermore, there is a growing body of evidence implicating infection during the perinatal and neonatal periods as an important contributor to an increased risk of adverse neurodevelopmental outcome in the preterm compared with the term population. In the neonatal period, because of the good visualization of cerebral parenchymal structures and ventricular system through the open fontanels, ultrasound still is the primary imaging modality of the neonatal brain. It allows also the depiction of enlarged subarachnoidal or subdural spaces, although not all of the convexity can be assessed. It easily demonstrates hemorrhagic or purulent components in the parenchyma, the ventricles, and parts of the extracerebral spaces, and can be repetitively used for follow-up. However, MR imaging better depicts white matter and cortical edematous or ischemic changes, delivers a complete assessment of the extracerebral spaces, and allows exact depiction of the posterior fossa, including brainstem, the latter two being almost blind spots for ultrasound. Besides delivering a complete anatomic coverage of the brain, MR imaging gives supplementary information by demonstrating contrast enhancement in inflammatory areas, or on tissue viability and metabolic processes by the use of diffusion-weighted imaging (DWI) and MR spectroscopy.
Bacterial infection
Epidemiology
Neonatal sepsis has been divided into early onset (within the first 7 days of life) and late-onset sepsis. Clinical signs for meningitis and early onset sepsis in the newborn are essentially similar and nonspecific. The incidence of early onset sepsis or meningitis is higher in infants weighing less than 1500 g compared with term neonates (15–19 vs 1–8 per 1000 live births), and is related to intrapartal infection from the maternal rectovaginal flora. Late-onset diseases present with a less fulminant course and meningitis is the most common presentation. During the last 10 years there has been an increased incidence of neonatal late-onset sepsis, predominantly caused by coagulase-negative staphylococci, and a decrease in incidence of early onset sepsis caused by group B streptococci (GBS). In early onset sepsis, the neonate can acquire the organism from his or her GBS-colonized mother through vertical transmission from the lower genital tract or from acquisition during passage through the birth canal. Risk factors that predispose a neonate to become infected are prematurity, lack of maternal anti-GBS IgG protection, placental infections with septic emboli, prolonged rupture of the fetal membranes, and high inoculum of the organism in the maternal anogenital tract. In some cases, direct or iatrogenic contamination of the CNS may occur (eg, after ventricular shunting). Maternal antepartal antibiotics have dramatically reduced the risk of early onset GBS infection in newborns by more than 80%. A corresponding change in Escherichia coli meningitis has not occurred and gram-negative bacillary meningitis still carries a worse prognosis than meningitis with a gram-positive organism. Maternal antibiotics may also have increased antibiotic resistance in E coli isolates from early onset bloodstream infections. Recent studies found no outcome difference between early onset GBS meningitis and gram-negative meningitis, but early onset GBS meningitis still has a worse outcome that late-onset GBS meningitis.
Pathophysiology
Meningitis complicates 5% to 20% of the cases of early onset neonatal sepsis. CNS damage is the result of a cascade of events, which begins with an extensive inflammatory vasculitis that may be followed by small or large vessel obstruction and infarctions. This vasculitis, which enables bacteria to cross the blood–brain barrier, takes place primarily at the level of the choroid plexus leading to plexitis and ventriculitis, with consecutive dissemination into the cerebrospinal fluid (CSF). Although in the strictest sense ventriculitis accompanies all cases of meningitis, it has been suggested that ventriculitis may occur as a primary process. After CSF dissemination, bacteria gain access to the leptomeningeal space and induce arachnoiditis. Through this inflammatory-destructive process, bacteria eventually invade the brain parenchyma, followed by cerebral tissue liquefaction and encephalomalacia. At the same time, inflammation of the arachnoid leads to adhesions and consecutive hydrocephalus development. However, the major element that contributes to an increase in intracranial pressure during bacterial meningitis is the development of cerebral edema, which may be vasogenic, cytotoxic, or interstitial in origin. Vasogenic cerebral edema is a consequence of increased blood–brain barrier permeability. Cytotoxic cerebral edema results from swelling of the cellular elements of the brain, most likely caused by the release of toxic factors in bacterial meningitis. Interstitial edema reflects obstruction of flow in normal CSF pathways, as in hydrocephalus. In neonates, cerebral edema is often the presenting clinical manifestation, because of rapid disease progression and higher vulnerability of the immature brain to the infectious agent. Nevertheless, distensibility of neonates’ cranial vault reduces the risk of fatal complication in case of brain herniation, which occurs in approximately 6% of acute meningits.
The pathophysiology of GBS meningitis varies according to age of onset. In early onset disease, autopsy studies demonstrate little or no evidence of leptomeningeal inflammation, despite the presence of abundant bacteria, vascular thrombosis, and parenchymal hemorrhage. By contrast, infants with late-onset disease usually have diffuse purulent arachnoiditis with prominent involvement of the base of the brain. These differences reflect the immaturity of the host immunologic response in the immediate neonatal period. A prominent sign of leptomeningeal inflammation is fluid accumulation in the subarachnoid or subdural space. Subdural effusions, which are initially sterile and mainly caused by toxin-induced increased permeability of the capillaries and veins of the internal layer of the dura mater, are rare in neonates and sometimes not easy to distinguish from enlarged subarachnoid spaces. True subdural purulent effusion and empyema or intraventricular pus accumulation whether caused by ventriculitis or recirculation of subarachnoid purulent collections are very rare in neonates.
After bacteria have gained access to brain parenchyma, focal purulent complications and brain abscesses may develop. Intracerebral abscesses complicating meningitis are most commonly related to Citrobacter , Proteus , Pseudomonas , Serratia , and Staphylococcus aureus . Gram-negative organisms and especially members of the Citrobacter and Proteus genus were reported to be the causative organism in most of the children younger than 1 month, whereas various Streptococcus species were the most common isolates in children older that 1 month. In this setting, hematogenous dissemination is the major source of origin in neonates, whereas contiguous infections (from sinusitis or otitis) and penetrating injuries are almost exclusively seen in older children. It must be emphasized that bacteria associated with brain abscess are those that cause meningitis and severe vasculitis, with only rare exceptions. Neonatal brain abscesses are characteristically large at the time of diagnosis, often multiple and typically lacking a complete capsule, which favors rapid enlargement. In more than 80% of cases, they are located in the cerebral hemispheres, especially frontal and parietal lobes, usually originating in the periventricular white matter, whereas the subcortical white matter and basal ganglia are more common locations in older children. Deeply located abscesses may rupture into the ventricular system. There is a continuum from focal cerebritis to mature abscess, a process that takes place during a period of 2 to 3 weeks, although it may be as short as a few days in neonates. Early cerebritis is characterized by intensive vasculitis, local inflammation, petechial hemorrhages, and surrounding edema, followed by necrosis of the center with irregular peripheral inflammatory tissue in later cerebritis stage. This host response builds the peripheral capsule of the abscess, which is initially poorly defined, thicker on the cortical side, and thinner on the medial or ventricular side, probably related to differences in vascularization. This difference may explain why abscesses predominantly rupture into the ventricular system, a sometimes fatal complication responsible for sudden death. In the late-capsule stage, a five-layer structure is recognizable at histology, with a thick granulation circumferential reaction and extensive peripheral edematous changes.
Long-term pathologic changes are mainly related to gliosis and encephalomalacia with variable amount of scarring and atrophy of the cortex and white matter, and hydrocephalus complications.
Neuroimaging
Imaging findings in acute neonatal bacterial meningitis vary greatly with the stage of infection. Extracerebral spaces and ventricular system may be normal, enlarged, or show evidence of protein or pus accumulation. Cerebral parenchyma alike may show normal signal intensity, evidence of edema, ischemia, infarction, hemorrhage, or necrosis with abscess and central purulent accumulation. Because of brain immaturity and poor immunologic response capabilities of the newborn in the immediate postnatal period, the physiopathologic cascade is not strictly timed and severe edema or abscess formation may be the most prominent or even the single presenting neuroimaging finding. Because of the lack of radiation, its higher sensitivity for ischemic lesions and infarctions, and its ability to easily detect purulent collections, MR imaging is the preferred imaging modality over CT, even in an emergency situation. In all cases, MR imaging scans must include pre–contrast- and post–contrast-enhanced T1-weighted and T2-weighted images in at least two perpendicular planes. Fluid attenuated inversion recovery (FLAIR) sequence and DWI are both recommended in this situation because of their high sensitivity in detecting purulent collections.
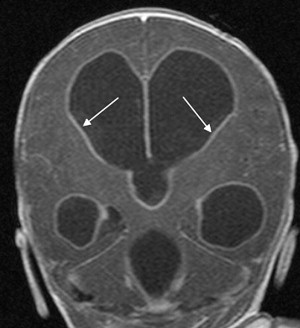
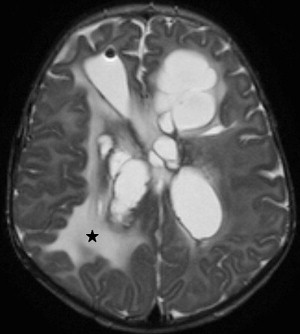
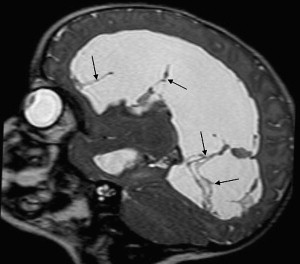
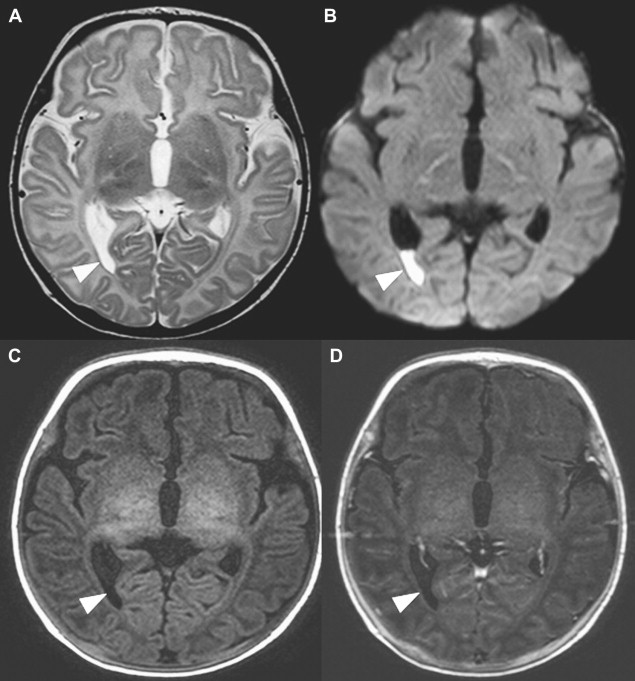
Choroid plexus infection and ventriculitis are often detected together, and show a characteristic triad consisting of choroid plexus engorgement, ependymal lining contrast enhancement, and intraventricular dependant debris and pus accumulation mainly in the occipital horns ( Fig. 1 ). Periventricular edema may be related to bacteria-induced periventricular white matter necrosis or to unbalanced CSF circulation with development of hydrocephalus ( Fig. 2 ).
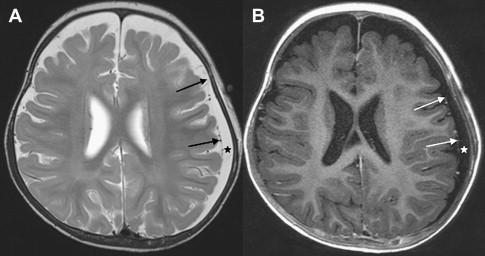
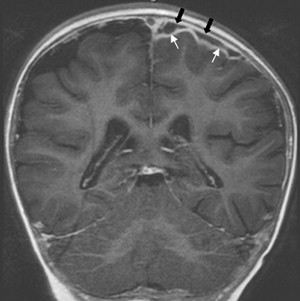
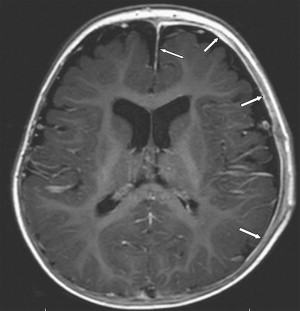
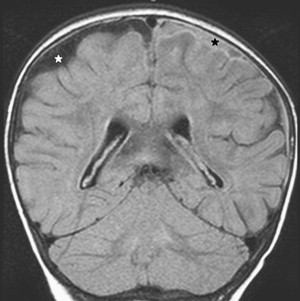
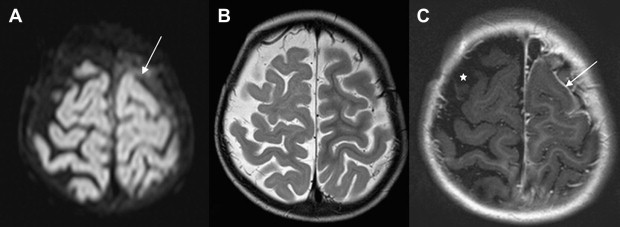
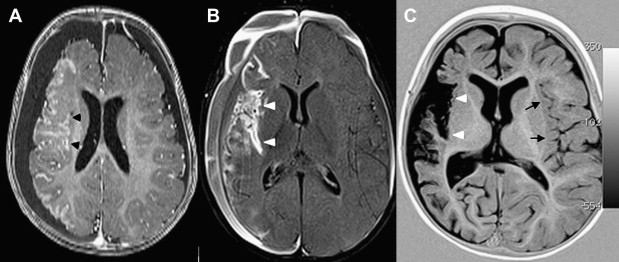
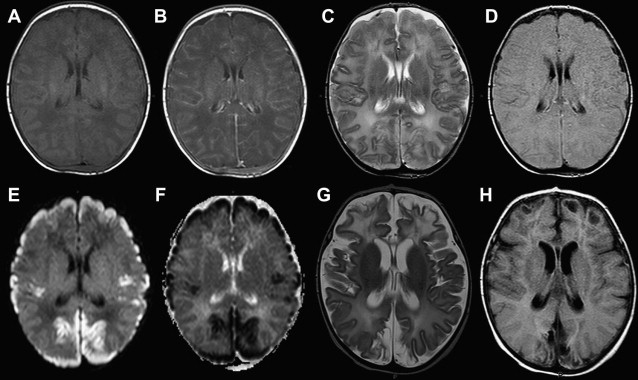
Once inflammation has reached the CSF compartment, arachnoiditis is the natural next step in the cascade of events. Nevertheless, the initial signs of involvement of the meninges, such as enlargement of the subdural or subarachnoid space, are nonspecific and may go unrecognized ( Fig. 3 ). However, contrast enhancement of the arachnoidea may be completely lacking in the natural history of GBS meningitis of the newborn. When present, contrast enhancement is seen as a pencil-shaped line covering the gyri and sometimes seen extending within the depth of the sulci ( Fig. 4 ). Contrast enhancement of the dura, however, is seen as a thickened line underneath the calvarium opposite to the arachnoid membrane and separated from it by the subdural space, and may persist several months after complete clinical recovery ( Fig. 5 ). Adhesions may occur everywhere within the CSF and consist of enhancing fibrin webs in the subarachnoid, subdural, and even intraventricular spaces ( Fig. 6 ). Before true purulent accumulation occurs, the intense inflammatory reaction produces cellular debris from leuocytic infiltrates and fibrin or protein residues, which can be seen as high signal intensity change of the CSF-filled spaces on FLAIR imaging ( Fig. 7 ), but may be difficult to recognize on T1-weighted, T2-weighted, or DWI ( Fig. 8 ). DWI is best at demonstrating the presence of truly purulent accumulation, which shows very high signal intensity, because of the higher viscosity of these collections ( Fig. 9 ). It must be emphasized that purulent collections may be completely missed on T1- or T2-weighted imaging ( Fig. 10 ).
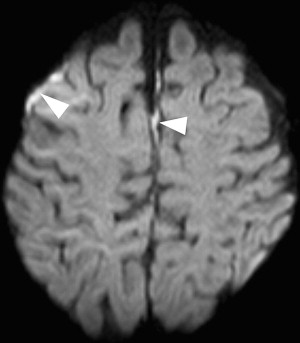
Large territorial or small lacunar peripheral infarctions result from either primary involvement of the vessels by way of blood-borne bacteria, or secondary caused by extension of the infection along the perivascular subpial spaces with inflammation of the walls of the arteries (arteritis). Vascular complications may also occur because of a breakdown of the blood–brain barrier. It may also induce cortical pseudolaminar necrosis, which appears as T1- and T2-hyperintensity and shows contrast enhancement ( Figs. 11 and 12 ).
Venous involvement in meningitis is rare. It mainly expresses itself as thrombophlebitis and venous thrombosis, a complication usually seen in combination with dehydration. On MR imaging, venous thrombosis displays itself as T1-isointensity to -hyperintensity of the thrombus within the vessel, depending on the age of the clot. Known complications of venous thrombosis are hemorrhagic transformations, which involve the matching venous drainage area.
Last in the chain of events in meningitis, brain parenchymal involvement leading to focal cerebritis produces an ill-defined area of T1-hypointensity and T2-hyperintensity on MR imaging. Mass effect is moderate and best identified through narrower sulci compared with the other side. Faint contrast enhancement may be detected within the edematous area. There is a continuum in MR imaging findings from early cerebritis to full-blown late-capsule stage abscess, with a continuously stronger demarcation of the purulent center against a peripheral initially incomplete, later complete inflammatory capsule. This last stage consists of a highly T1-hypointense and T2-hyperintense center, surrounded by a marked closed ring that consistently shows strong contrast enhancement. Peripheral edema is present to a variable amount in all stages ( Fig. 13 ). The presence of hemorrhagic foci is revealed by focal T1-hyperintensity on pre–contrast-enhanced imaging. In analogy to the previously described extracerebral purulent fluid, DWI is highly sensitive in showing central pus accumulation in late-stage abscess, because of the high viscosity of the fluid ( Fig. 14 ). Markedly reduced diffusivity (bright on DWI and dark on apparent diffusion coefficient [ADC] maps) is characteristic and can be used as a surrogate marker of treatment response. MR spectroscopy has been used to analyze the central components of abscesses, revealing first the absence of normal brain metabolites ( N -acetylaspartate, choline, and creatine) and second the presence of residues from anaerobic glycolysis (lactate, lipids, acetate, and succinate) and proteolysis (aminoacids) ( Fig. 15 ).

Related posts:
 Neonates with Seizures: What to Consider, How to Image
Neonates with Seizures: What to Consider, How to Image
 MR Imaging of the Newborn
MR Imaging Workup of Inborn Errors of Metabolism of Early Postnatal Onset
MR Imaging of the Newborn
MR Imaging Workup of Inborn Errors of Metabolism of Early Postnatal Onset
 Congenital Cardiac Defects and MR-Guided Planning of Surgery
Congenital Cardiac Defects and MR-Guided Planning of Surgery
 Birth-Related Injury to the Head and Cervical Spine in Neonates
MR Imaging of the Neonatal Musculoskeletal System
Birth-Related Injury to the Head and Cervical Spine in Neonates
MR Imaging of the Neonatal Musculoskeletal System
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


