Chapter Outline
Normal and Abnormal Neonatal Bowel Gas
Jejunal and Ileal Stenosis and Atresia
This chapter discusses gastrointestinal malformations that are manifested during the neonatal period. Some are grossly apparent at birth (e.g., gastroschisis, omphalocele, diaphragmatic hernia); others usually are manifested within the first hours or days of life (e.g., esophageal, small bowel, or colonic atresia; meconium ileus; meconium plug; Hirschsprung’s disease). Many of these diagnoses can be suggested by prenatal ultrasound.
Rotational Anomalies
Embryology
During the sixth gestational week, rapid elongation of the midgut and hindgut results in their herniation into a sac in the midline of the anterior abdominal wall. Before returning to the peritoneal cavity during the ninth week of gestation, the midgut revolves 90 degrees around the superior mesenteric artery. Once it is within the abdominal cavity, the bowel rotates an additional 180 degrees, which positions the duodenojejunal junction to the left of the spine at the level of the stomach, the jejunum in the left upper quadrant, and the ileum in the right hypochondrium or right lower quadrant. The colon undergoes a separate counterclockwise rotation of 270 degrees, which brings the cecum into the right lower quadrant. This normal rotation is associated with a broad mesenteric base, extending from the left upper quadrant to the right lower quadrant. Attachments at two sites keep the bowel fixed in proper position: the ligament of Treitz at the duodenojejunal junction and an attachment at the cecal base.
Deviation from normal rotation and fixation occurs universally in children with omphalocele, gastroschisis, and diaphragmatic hernia. Variations of rotation may also be present in children with asplenia and polysplenia syndromes, duodenal stenosis or atresia, and Hirschsprung’s disease. However, malrotation frequently exists as an isolated anomaly.
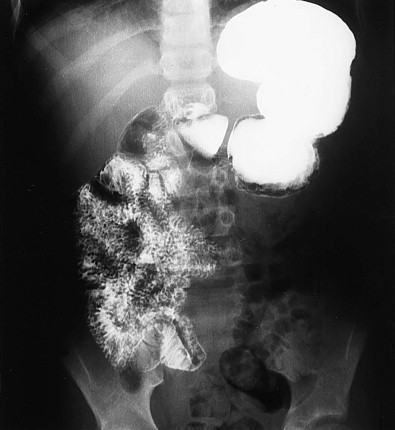
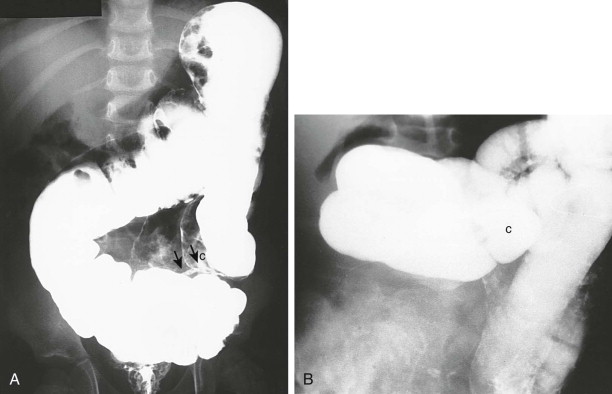
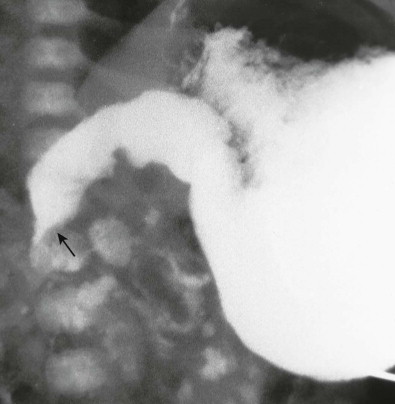
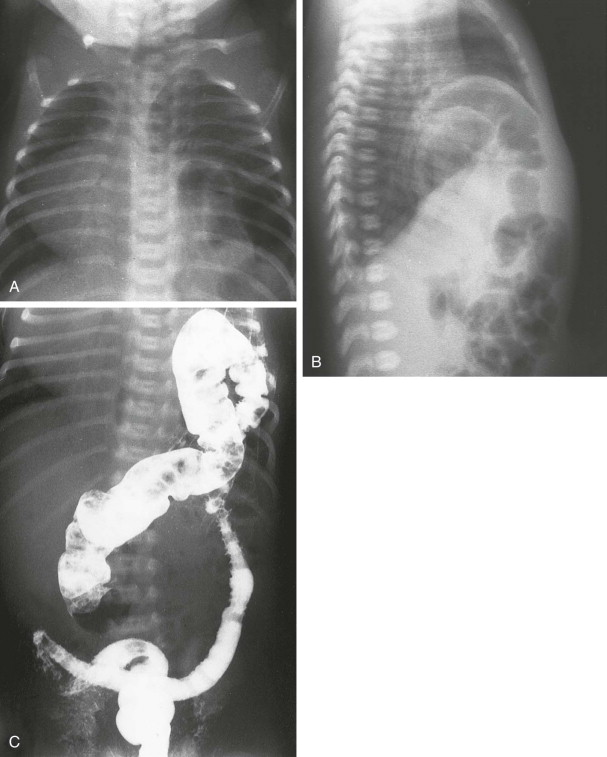
The spectrum of rotational abnormalities is broad ( Figs. 114-1 to 114-3 ). In some children, the process of rotation takes place, but fixation fails to occur. In others, there are only minor variations from the normal position. Complete nonrotation is said to be present when the jejunum is to the right of the spine and the ileum is in the pelvis or to the left of the spine. Most clinical problems arise in children with the greatest deviation from the normal rotational pattern. In classic malrotation, the cecum lies in the midabdomen or left of the midline (see Fig. 114-2 ) and may be fixed in place by broad bands that emanate from the undersurface of the liver. These Ladd bands cross the duodenum and may cause extrinsic compression and obstruction of the gut at this level (see Fig. 114-3 ).
Clinical Findings
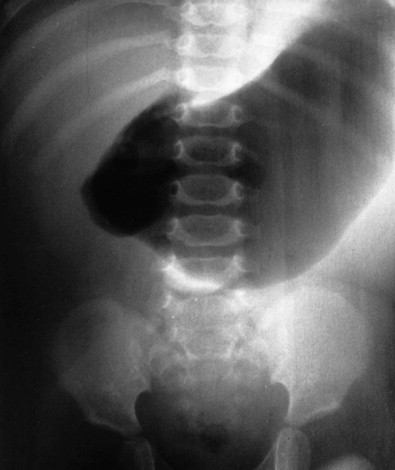
Most children with malrotation present in the first few months of life. They have acute abdominal symptoms if there has been acute volvulus with twisting of the bowel on its shortened mesentery or chronic vomiting and failure to thrive because of the obstructing Ladd bands (see Fig. 114-3 ). Midgut volvulus may produce vascular compromise, which can lead to gangrene of the entire small bowel if it is not promptly diagnosed and treated ( Fig. 114-4 ).
In some children, malrotation is not detected until later, when studies are done for other purposes. Uncommonly, malrotation can be associated with chronic volvulus. This condition interferes with lymphatic and venous drainage, which produces malabsorption or failure to thrive. Acute volvulus with infarction of bowel does occur, although rarely, in older children and adults. Motility abnormalities may persist after corrective surgery. Other duodenal abnormalities occur frequently in children with malrotation: duodenal atresia, annular pancreas, and preduodenal portal vein.
Radiologic Finding s
Abdomen radiographs are of little value in the child with an uncomplicated rotational abnormality because the positions of the duodenojejunal junction and cecum are rarely definable without positive contrast media. Sometimes, abnormal configuration of the gas in the right hypochondrium (i.e., duodenal triangle) may suggest the diagnosis of malrotation with volvulus. Normal findings on radiographs do not exclude the diagnosis of malrotation with volvulus.
In the neonate or child with abdominal pain or vomiting, abdomen radiographs showing gaseous distention of the stomach and duodenal bulb suggest a high obstruction (see Fig. 114-4 ). This appearance (i.e., double bubble) can also be seen in duodenal atresia and annular pancreas. A contrast study may be necessary for differentiation between the relatively benign complication of Ladd bands (see Fig. 114-3 ) and the surgical emergency of volvulus (see Fig. 114-4 ).
In the child with midgut volvulus, the abdominal gas pattern may be normal or show high or low obstruction, or the abdomen may be gasless. Because abdomen radiographs are usually nondiagnostic and the consequences of delayed diagnosis are grim, contrast studies should be performed emergently when volvulus is suspected.
The first study performed to detect the malrotation and its complications should be an upper gastrointestinal series. The barium enema can show a malpositioned or malfixed cecum (see Fig. 114-2 ), but normal results of a barium enema do not exclude malrotation with volvulus. For this reason, an upper gastrointestinal series may need to be performed after the enema if a low obstructive lesion is not found and malrotation is still considered.
The diagnosis of malrotation can be made sonographically, although it is usually an incidental finding on studies performed for other reasons. Failure of the bowel to rotate normally produces an abnormal relationship between the superior mesenteric artery and vein. With volvulus, the superior mesenteric vein may wind around the superior mesenteric artery, inconstantly producing the whirlpool sign on ultrasound. Sonography may exclude malrotation by showing the third portion of the duodenum between the aorta and superior mesenteric artery. Computed tomography (CT) can also show these vascular changes and demonstrate malpositioned and twisted bowel.
Gastroschisis
General Considerations
Development of the anterior abdominal wall is complex, with the orderly ingrowth of four separate folds (i.e., cranial, caudal, and two lateral) necessary for normal closure. Gastroschisis is a parasagittal defect, usually to the right of the normally positioned and normal-appearing umbilical cord, through which bowel herniates into the amniotic fluid.
Clinical Findings
The herniated bowel has no covering membrane or sac and is therefore associated with a rise in the maternal serum α-fetoprotein level. Occurring in about 1 of 10,000 live births, gastroschisis can be diagnosed prenatally with sonography. At birth, the defect and herniated bowel are apparent and not easily confused with other abdominal wall defects.
Malrotation or nonrotation of the bowel is the rule in gastroschisis, but it rarely leads to complications. Bowel atresia, present in 20% of cases, is usually the only anomaly, but it is an important factor contributing to postoperative morbidity.
Treatment
Even though a small amount of bowel usually is herniated through a small defect, surgical repair is associated with a postoperative mortality rate of 5% to 25%, with major complications of sepsis and electrolyte problems. Antenatal exposure to amniotic fluid produces bowel wall edema and inflammatory thickening of the serosa, which interferes with peristaltic function, even after repair. In utero, the exposed bowel and mesentery may become shortened and coiled, which also affects postnatal function. Short gut syndrome may decrease intestinal absorption and cause diminished constitutional growth. Children with short gut or hypoperistalsis can be supported with parenteral hyperalimentation, but this also can create management problems: venous thrombosis, liver disease, and cholelithiasis. Cesarean section, once routine in an attempt to diminish the intrauterine bowel changes, has been shown to produce no difference in postnatal and postoperative bowel function.
The type of surgical correction, primary or delayed, depends on the size of the defect and the presence of other complications, such as atresia and short gut. In addition to covering the herniated bowel, with placement of an abdominal silo, it may be necessary to create a stoma to decompress the dilated bowel proximal to an atresia.
After surgery, motility changes are universal in children with gastroschisis. The initial postoperative paralytic ileus is frequently followed by marked prolongation of intestinal transit. These children also have a high incidence of significant gastroesophageal reflux. Even though they are usually term infants, necrotizing enterocolitis (NEC) occurs in 23% of cases at 1 to 4 months after repair and may be manifested atypically; only 36% of affected children have blood-streaked stool.
Radiologic Findings
The prenatal sonographic diagnosis of gastroschisis is based on the observation of normal umbilical cord insertion in a fetus with an anterior abdominal wall defect through which bowel has herniated. No membrane covers the bowel; if fetal ascites or a covering membrane is present or if the liver is detected in the herniated viscera, omphalocele is the more likely diagnosis. Thickening of the exteriorized bowel loops strongly suggests gastroschisis. Amniotic fluid volume is usually normal. A bowel caliber of more than 17 mm suggests that an atresia is present; similarly, bowel of a smaller diameter is usually associated with bowel continuity.
Abdomen radiographs show the normally positioned umbilical clamp separated from herniated bowel loops, which are outlined by air ( Fig. 114-5 ). Postoperative radiographs should be scrutinized to detect changes of NEC: ileus, dilated bowel loops, and intramural air. Postsurgical barium studies are used to detect gastroesophageal reflux, bowel loop dilation, obstructive adhesions, amount of bowel present, and abnormalities of position, peristalsis, or transit time.
Omphalocele
Clinical Findings
Omphalocele, present in about 1 of 5000 live births, is a midline defect of variable size through which bowel, liver, spleen, pancreas, and uterus may protrude. A membrane or sac usually covers the herniated organs, but the sac may be ruptured at birth. The umbilical cord inserts into the apex of the sac. The bowel is malrotated, and 8% to 20% of these children have Meckel’s diverticulum.
The diagnosis of omphalocele can be made with prenatal ultrasound. Maternal α-fetoprotein levels may be elevated, but because of the covering sac, they tend to be less than those found with gastroschisis. Associated anomalies are seen in about 50% to 80% of infants with omphalocele, including tetralogy of Fallot and atrial septal defect as well as other cardiac, central nervous system, and gastrointestinal anomalies. Detection of anomalies may influence the outcome or management of the pregnancy; certain anomalies are associated with fetal demise, and others may result in planned termination of pregnancy. Children with Beckwith-Wiedemann syndrome account for almost 12% of the population with omphalocele. These infants are large at birth and have a large tongue. A specific pancreatic abnormality, nesidioblastosis, predisposes the infants to hypoglycemia, even in the neonatal period. Down syndrome (i.e., trisomy 21), trisomy 13, and trisomy 18 are associated with an increased incidence of omphalocele.
Children with small anterior abdominal defects (e.g., gastroschisis, small omphalocele) tend to have a normally developed thorax. Those with giant omphaloceles (i.e., containing liver and bowel) have a small thorax and an increased incidence of pulmonary hypoplasia and respiratory insufficiency. They may require ventilatory support after surgery.
Surgical management may take many forms. In most children, the defect is corrected by primary skin closure or closure with a silo. The surgical approach is determined by the size of the defect, and larger defects may require a staged reduction.
Radiologic Findings
Prenatal sonographic diagnosis of omphalocele is based on visualization of the umbilical cord inserting into the membrane covering structures anterior to the abdominal wall of the fetus. Fetal ascites, abnormal amounts of amniotic fluid, and associated congenital defects are supportive ancillary findings. Thickening of exteriorized bowel loops or absence of membrane or sac suggests the diagnosis of gastroschisis.
Postnatal abdominal radiographs ( Fig. 114-6 ) depict the omphalocele as a soft tissue density whose margins are well defined by the adjacent air. Unlike gastroschisis (see Fig. 114-5 ), the bowel loops are not individually seen unless the omphalocele sac has ruptured. Malrotation of the bowel and malposition of other organs are identified on postoperative imaging studies.
Diaphragmatic Hernia
Foramen of Bochdalek Hernia
Embryology
In early fetal life, the peritoneal cavity and the pleural space are in continuity. At week 8, just before anteriorly herniated bowel returns to the abdominal cavity, the communication between these two spaces is closed by the development of the diaphragm. If the bowel returns to the abdomen prematurely or if the diaphragm develops late or incompletely, a diaphragmatic hernia develops. Affected children have malrotation of the bowel because the normal rotation that occurs as the bowel returns to the abdomen is interrupted. A membrane covers the herniated gut in only 10% of Bochdalek hernias.
Clinical Findings
The posterolateral or Bochdalek hernia occurs in approximately 1 of 3000 live births. Some diaphragmatic hernias may be acquired after trauma or infection. In the neonatal period, diaphragmatic hernias can develop in association with group B streptococcal infection. Late presentation or detection of an asymptomatic diaphragmatic hernia has also been described.
Congenital diaphragmatic hernia occurs six to nine times more often on the left, presumably because the pleuroperitoneal canal closes earlier on the right; about 3% of children have bilateral diaphragmatic hernia. Left-sided hernias usually contain portions of the gastrointestinal tract. The liver may extend into the thorax with left or right diaphragmatic hernias. Regardless of the side on which the hernia occurs, the ipsilateral and contralateral lungs are compressed, and the most critical factor in determining the neonate’s outcome is the degree of pulmonary hypoplasia.
Diaphragmatic hernia should be suspected clinically when a neonate with severe respiratory distress has a scaphoid abdomen. Because bowel contents are in the thorax, the abdomen lacks its normal protuberant appearance. Most newborns with diaphragmatic hernia are rapidly intubated and resuscitated; when they are stable, they undergo surgical repair. All children are carefully inspected for midline defects (i.e., cleft lip and palate, spina bifida, and omphalocele) and cardiac lesions (i.e., ventricular septal defect and tetralogy of Fallot) because associated anomalies also determine outcome.
Despite ventilatory support and pharmacologic manipulation including inhaled nitric oxide to correct pulmonary hypertension, a common complication of diaphragmatic hernias, early presurgical mortality rates range from 20% to 80%. To improve survival, extracorporeal membrane oxygenation (ECMO) is used in infants who experience significant respiratory insufficiency resulting from pulmonary hypoplasia. ECMO may be performed before, during, or after surgery. This lung bypass system entails placement of large-bore cannulas into the aortic arch through the right common carotid artery and the right atrium (venoarterial approach) or a single double-lumen catheter into the right atrium (venovenous approach) through the right internal jugular vein. Heparinization is routine. The lungs are minimally ventilated and given the chance to mature and grow. ECMO has increased postsurgical survival to more than 80%, but it is not without complications: bleeding at the neck wounds, intracerebral and pleural hemorrhage, and difficulty with later central line placement. Children who require ECMO have a poorer neurologic outcome than those who do not. Because these children are sicker from the moment of birth, the neurologic deficits have a multifactorial cause, including ECMO.
The fact that many infants with diaphragmatic hernias still died before undergoing repair drove the research in prenatal surgical intervention throughout the 1990s. The observed-to-expected fetal lung volume measured by magnetic resonance imaging (MRI) is reported to be a predictor of survival. In fetuses with a very low observed-to expected fetal lung volume, one fetal surgical technique employed involves fetal tracheal occlusion; this causes fluid to fill the lungs and encourages their expansion despite the extrinsic pressure produced by herniated abdominal contents. The clip or balloon occluding the trachea is removed at the time of birth, allowing normal respiration. Results with this technique, used only in those whose prenatal ultrasound indicates severely compromised lung parenchyma, have been promising.
Radiologic Findings
The prenatal sonographic diagnosis of diaphragmatic hernia is made when the heart and other mediastinal contents are shifted from the midline and a “mass” (i.e., liver or gut) is present in the thorax. A fetal abdominal circumference below the fifth percentile correlates with a poor prenatal and postnatal course in some studies. Fetal ascites, pleural effusion, and polyhydramnios have been associated with diaphragmatic hernia.
Prenatal ultrasound and MRI have documented the presence of a portion or all of the liver in many diaphragmatic hernias. When it is not diagnosed prenatally, the affected infant presents with respiratory distress shortly after birth. Abdomen radiographs can determine the amount of bowel present in the abdomen. Chest radiographs can exclude other causes of neonatal respiratory distress: pulmonary immaturity, congenital pulmonary airway malformation, and pneumonia. Complications of resuscitation and other congenital abnormalities that may affect management should be sought. Pneumothorax is common and requires prompt treatment.
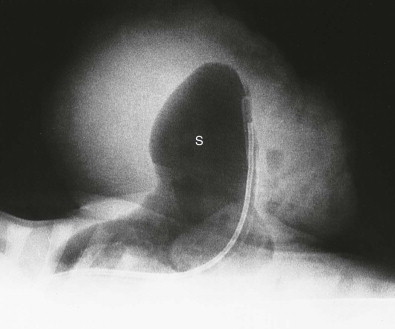
On early radiographs, the herniated bowel loops may appear as a soft tissue mass. With time and air swallowing, the chest radiographs have a more typical bubbly appearance ( Fig. 114-7 ). Diaphragmatic hernias that contain liver may appear more solid and may be accompanied by a pleural collection. On occasion, mild diaphragmatic hernias are not diagnosed until later in childhood.
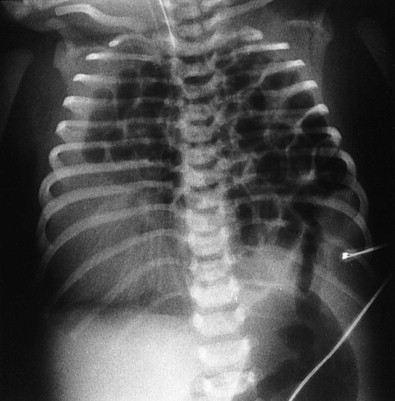
If the nature of the thoracic contents is uncertain, a nasogastric tube should be inserted to define the stomach and to introduce air into the bowel. Contrast studies are rarely needed to visualize the upper or lower gastrointestinal tract before surgery ( Fig. 114-8 ).
The mediastinum remains shifted to the side opposite the hernia on immediate postoperative chest radiographs. The ipsilateral lung appears as a soft tissue density outlined with air close to the mediastinum. The ipsilateral pneumothorax is expected and may simulate tension pneumothorax because of the long-standing mediastinal shift. Attempts to evacuate this pneumothorax may result in overexpansion of the hypoplastic contralateral lung and cause contralateral pneumothorax. During the first few postoperative days, as the ipsilateral lung expands, fluid may occupy a portion of the pleural space and produce haziness on that side of the thorax.
Before a child is placed on ECMO, cardiac, cranial, and abdominal ultrasound is performed to identify those who can safely receive this treatment. Neonates with intracranial hemorrhage or with a lethal anomaly are excluded from ECMO.
Neonates treated with ECMO receive a daily chest radiograph. Evaluation of line placement is necessary; kinking and dislodgment of support lines may occur and compromise treatment. Lung opacity is often diffusely increased because of the planned underventilation, increased fluid in the lungs, and, uncommonly, bleeding. Pleural hemorrhage develops in about 30% of children on ECMO and may be manifested as a typical pleural collection or, when it is associated with pneumothorax, an unexpected mediastinal shift. The esophagus may appear as a mediastinal air or fluid mass in the midline because it frequently dilates from gastroesophageal reflux. Although the replacement of the bowel loops into the abdomen usually is well tolerated, about 20% of long-term survivors experience intestinal obstruction, and about 13% require surgery for relief of the obstruction.
The most striking finding on the chest radiographs of many long-term survivors is the degree to which the hypoplastic lungs may grow and develop. In many children, a chest radiograph at age 2 or 3 years has a nearly normal appearance.
Foramen of Morgagni Hernia
Herniation of colon or other abdominal structures into the retrosternal space is rare and occurs when the anteromedial aspect of the diaphragm develops abnormally. The so-called foramen of Morgagni hernia accounts for about 2% to 4% of all diaphragmatic hernias.
Respiratory and gastrointestinal complaints are common but not necessarily related to the hernia. The child may be asymptomatic, and the abnormality may be detected on a chest radiograph obtained for a nonrelated purpose. Only 50% are detected by age 5 years.
Although the amount of bowel herniated into the chest in Morgagni hernias is usually much less than in Bochdalek hernias, malrotation or malfixation is common. In contrast to Bochdalek hernias, Morgagni hernias usually are right sided and have a covering or sac.
The differential diagnosis of air-containing Morgagni hernias includes pneumonia, atelectasis, pneumatocele, abscess, and congenital pulmonary airway malformation. If the liver is herniated, the solid appearance may simulate a tumor of the diaphragm, a pericardial mass, or an anterior mediastinal mass. Morgagni hernias are corrected even in asymptomatic children because of the potential for incarceration and strangulation.
Radiologic Findings
Anteroposterior chest radiographs may show a soft tissue opacity or air along the heart border ( Fig. 114-9 ). On the lateral projection, the anterior location of the hernia and visualization of bowel establish the diagnosis. In some cases, additional imaging may be needed to identify the nature and extent of the hernia (see Fig. 114-9 ).
Normal and Abnormal Neonatal Bowel Gas
With the first breath, the neonate begins to aerate the respiratory and gastrointestinal tracts. Unless there is obstruction, these processes tend to parallel each other. Several problems delay the passage of air into the gastrointestinal tract. The most obvious is a mechanical obstruction in the oral cavity or esophagus, most often esophageal atresia. Drugs given during labor and delivery can depress the swallowing mechanism, diminish the amount of air swallowed, and delay passage of air distally into the small bowel and colon.
Abdomen radiographs that are initially normal but then become gasless raise the possibility that the neonate has sepsis, has developed electrolyte imbalance, is receiving gastric suction, or is being paralyzed while being ventilated. These processes decrease the amount of air swallowed and the amount available for passage into distal bowel.
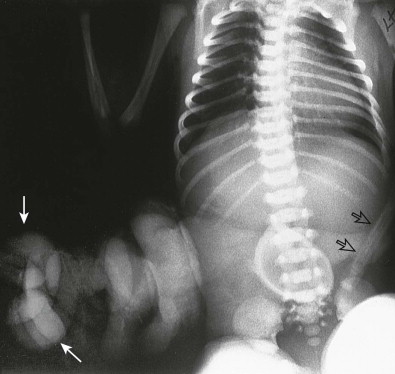
The bowel gas pattern should be carefully scrutinized in any neonate with respiratory or gastrointestinal symptoms. Gas should be symmetrically distributed throughout the abdomen in a mosaic pattern. Paucity or malposition of the gas may confirm a suspected diaphragmatic hernia. An abdominal mass may displace bowel loops and, through pressure on the diaphragm, cause respiratory distress. With a high bowel obstruction, a few loops of bowel in the upper abdomen are distended with air and fluid ( Fig. 114-10 ). With distal obstruction, dilated bowel loops fill the abdomen ( Fig. 114-11 ).
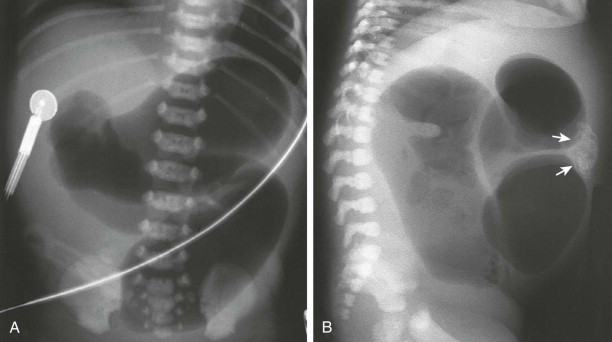
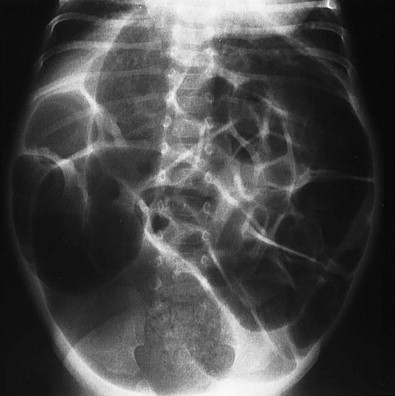
In the neonate, it may be difficult to differentiate small from large bowel because haustra are poorly developed. Although location may allow differentiation of small from large bowel, distended small bowel may fill the space usually occupied by the colon or even simulate a distended stomach.
It is important to determine the most distal extent of air because the differential diagnosis depends on the level of the obstruction. Additional views may be necessary to detect air within the colon. Prone lateral rectal views may be useful if an extremely distal lesion is suspected. A contrast study of the colon may be performed to determine if there is an obstruction or to identify the level and nature of the obstruction.
Abdominal Masses
Clinical Findings
The differential diagnosis of abdominal mass in the newborn is extensive: ovarian cyst or tumor, alimentary tract duplication, mesenteric or omental lymphangioma, cyst or tumor of the spleen or liver, choledochal cyst, cystic meconium peritonitis, hydrometrocolpos, and retroperitoneal masses. Most neonatal abdominal masses originate in the kidneys; ureteropelvic junction obstruction and multicystic dysplastic kidneys are the most common causes. Gastrointestinal lesions account for 8% to 15% of neonatal abdominal masses.
The mass may be detected by prenatal sonography or by abdominal palpation after birth. In some neonates, the mass is sufficiently large to distend or distort the abdominal wall and cause respiratory distress. Ascites may be present or simulated. Pain or obstruction results if the mass is producing pressure on an adjacent structure or if there is torsion of the mass.
A bruit over the liver in an infant with congestive heart failure suggests a hepatic hemangioma. If a mass is present over the buttocks, the abdominal mass may represent internal extension of a sacrococcygeal teratoma.
Radiologic Findings
Abdomen radiographs may show obstruction or displacement of bowel, relatively nonspecific findings. Calcifications suggest meconium peritonitis, teratoma, and hepatoblastoma; spinal anomalies suggest anterior sacral meningocele, obstructed cloacal deformity, and sacrococcygeal teratoma with an internal component.
Ultrasound is the premier imaging technique for abdominal and pelvic masses in neonates ( Fig. 114-12 ). The following questions should be addressed:
From what compartment or organ does the mass originate?
Is the mass solid, cystic, or septate?
Is there a wall, membrane, or capsule around it?
Do the structures surrounding the mass look normal?
Is there ascites?
Are there any sites of spread or extension within the abdomen or retroperitoneum?
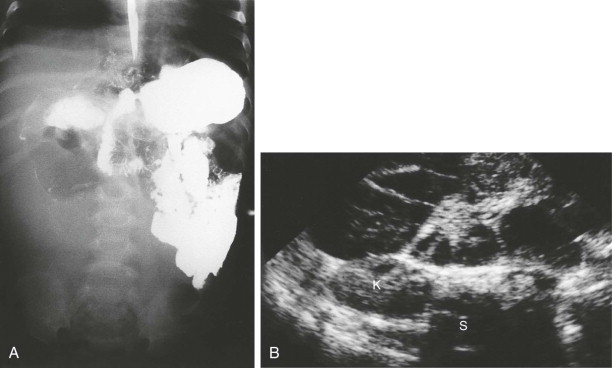
On the basis of the information obtained, the child may proceed to surgery, undergo further radiologic evaluation, or be observed and restudied after a period of observation.
Necrotizing Enterocolitis
Clinical Findings
NEC is a life-threatening process that primarily affects the gastrointestinal tracts of premature infants. Signs and symptoms of NEC usually develop in the first 2 weeks of life: rising gastric residual volume, abdominal distention, bloody stools, lethargy, and even changing respiratory status.
Associations other than prematurity have been noted: bowel ischemia of any cause; abnormal gut hormones, immunoglobulins, or peristalsis; enteral feedings, especially high volumes of formula with high calorie concentration; and maternal cocaine use. The occasional epidemic nature of NEC indicates that a viral or bacterial agent may play a role in some cases.
Pathologic examination reveals ulceration that begins in the mucosa and extends to the submucosa; inflammatory cells may be present in multiple bowel layers. Pneumatosis intestinalis is seen in the submucosa and subserosa. In 50% of cases, normal areas of bowel are interposed between diseased segments. Inflammatory pseudomembranes occur in less than 10%. Many specimens concurrently show acute changes and reparative changes in the same bowel segment. Complications of these processes include gangrene, perforation with peritonitis or enterocyst formation or stricture formation, enteric fistulas, and sepsis.
Timing of surgery is crucial; ideally, surgery should take place when necrosis is present but before bowel perforation. Erythema of the abdominal wall or positive result of paracentesis suggests bowel perforation and mandates surgical treatment. Pneumoperitoneum is an indication for surgery. These clinical and radiologic signs clearly indicate that perforation has occurred.
Strictures are late findings that develop in about 9% to 35% of children with NEC. About 75% occur in the colon, usually in the region of the splenic flexure; 15% are multiple; and the terminal ileum is involved in 15% of affected infants. They can be manifested after a delay of 20 months, but when they are seen on early studies, they may resolve spontaneously. Children whose NEC-induced bowel perforations are treated with bowel diversion or percutaneous peritoneal drainage have a lower rate of stricture formation than do those who received only medical management. To exclude a stricture, children with NEC may undergo an antegrade contrast study of the entire gastrointestinal tract before feeding is resumed. In infants with diversions, the bypassed bowel should be studied to exclude a stricture before bowel continuity is reestablished. Medical management of infants with suspected NEC includes parenteral nutrition and antibiotic therapy.
Most children with NEC, treated medically and surgically, survive. The greatest mortality rate is seen for small premature infants who, too frail to undergo standard laparotomy, are treated with percutaneous peritoneal drainage. Late complications include short gut syndrome, sepsis, abdominal abscess, recurrent NEC, and stricture formation as well as some extragastrointestinal problems. NEC survivors constitute the largest group of children with short gut syndrome.
Radiologic Findings
Abdomen radiograph findings include gastric dilation, a persistently dilated bowel loop, or an unchanging bowel gas pattern. Pneumatosis intestinalis ( Figs. 114-13 and 114-14 ) is manifested later, and large collections of intramural gas create a linear streaky pattern that parallels the bowel wall or, when seen en face, is manifested as circular lucency around the bowel lumen. Although the bubbly appearance of pneumatosis intestinalis may suggest feces, the premature infant rarely has formed stool within the colon in the first 2 weeks of life.
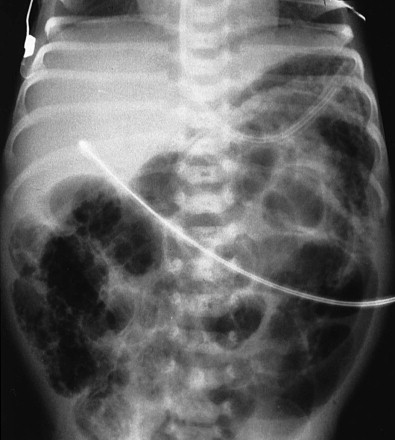
Gas enters the mesenteric veins and subsequently the portal vein and its branches, producing streaky lucencies radiating to the periphery of the liver (see Fig. 114-14 ). This is an evanescent sign in most children, but to many physicians, it indicates the need for surgery.
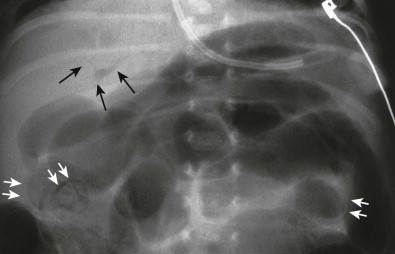
Bowel perforation, the most serious complication of NEC, is manifested by free intraperitoneal air. When the amount of air is large, a diffuse lucency appears over the liver or midabdomen. Air may outline the falciform ligament ( Fig. 114-15 ). The inner and outer surfaces of the bowel wall may be clearly seen. In a few children, air surrounds the umbilical arteries and produces an inverted V sign in the pelvis. Air in Morison’s pouch produces a triangular lucency.
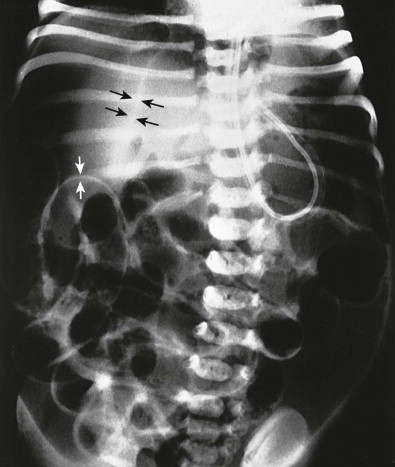
When free air is suspected, supine abdomen radiographs are supplemented by cross-table lateral radiographs, which are more easily obtained than decubitus or upright radiographs. Free air is visible as lucency anterior to the liver and stomach or as small, triangular lucencies projecting downward from the abdominal wall between the bowel loops.
Although ultrasound is not the primary imaging technique for NEC, it can show mural thickening of affected loops and portal venous gas before their detection on radiographs. Intrahepatic portal venous gas is seen as bright reflectors bubbling through the liver. The hepatic parenchyma develops unusually bright echoes in a patchy distribution. Severely affected (gangrenous) bowel loops may demonstrate diminished or absent blood flow when color Doppler imaging is used. Focal fluid collections, echogenic free fluid, increased bowel wall echogenicity, and increased bowel wall thickness are also ultrasonographic signs predicting poor outcome. Early diagnosis of NEC prompts therapy that in most cases aborts the process. When NEC is suspected clinically, the infant is treated accordingly, even in the absence of radiographic findings. If confirmation is required, the neonate can undergo upper gastrointestinal imaging with low-osmolality contrast agents or even contrast enema. A novel approach to confirming NEC employs CT. Urine specimens collected from neonates with NEC who have ingested diluted low-osmolality contrast material have a significantly higher mean CT attenuation value than the urine of normal infants.
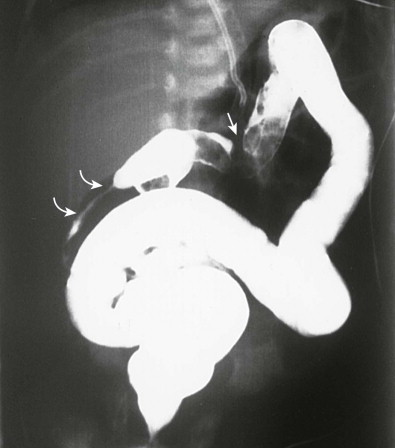
Strictures that can develop in the natural history of NEC can cause clinical and radiographic signs of bowel obstruction. Contrast enema ( Fig. 114-16 ) should be used to evaluate the entire colon and terminal ileum. Although some of the strictures may spontaneously regress, most are resected surgically or dilated with balloon catheters.
Esophageal Atresia
General Considerations
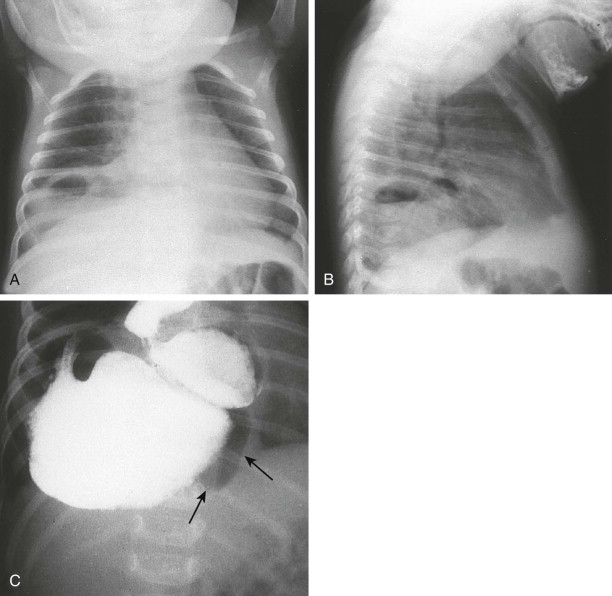
Esophageal atresia (EA) occurs in about 1 in 5000 live births; boys and girls are equally affected. In more than 80%, the trachea is connected to the distal esophagus by a congenital tracheoesophageal fistula (TEF), but in about 10%, the EA is complete ( Fig. 114-17 ). About 3% to 4% have a proximal fistula (with or without the distal fistula), and about 5% have no atresia but have an aberrant connection between the trachea and esophagus (i.e., H-type fistula).
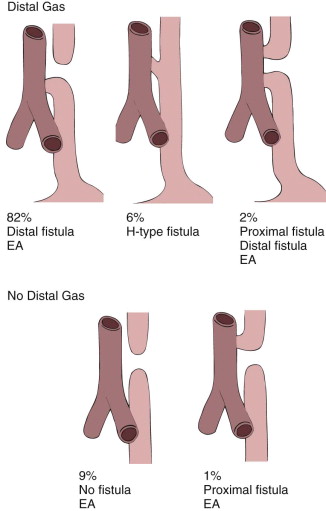
EA can be diagnosed prenatally but is more often diagnosed at birth, when the infant has difficulty handling secretions, or with the first feeding, when the infant has respiratory distress. Attempts to pass a nasogastric tube are usually unsuccessful; rarely, the tube may enter the trachea and pass into the TEF and distal esophagus. Prompt diagnosis of EA is necessary to protect the lungs.
The neonate with EA or with EA and TEF must be evaluated for exclusion of other anomalies, including trisomy 21 and the VATER association. The acronym VATER or VACTERL emphasizes that v ertebral, a norectal, c ardiac, t racheo e sophageal, r enal, and l imb anomalies may occur together. Physical examination and imaging studies are performed before surgery to find other anomalies that need emergent correction (e.g., duodenal atresia), influence the standard surgical correction of TEF (e.g., right aortic arch), or affect mortality (e.g., renal agenesis, duct-dependent cardiac lesions). In about 50% of infants, EA is an isolated anomaly.
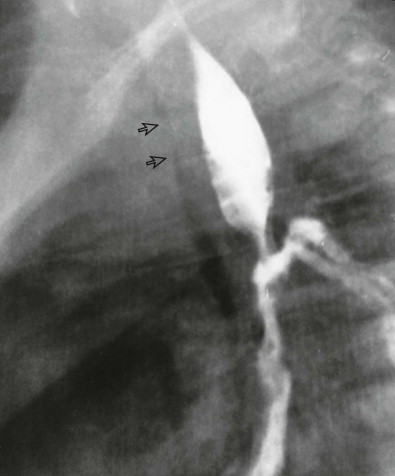
The presence of a distal fistula can be diagnosed clinically (see Fig. 114-17 ) because the affected neonate has a rounded abdomen and bowel sounds. The infant with EA and no distal fistula has a scaphoid abdomen and absent bowel sounds.
The stable neonate with EA and TEF undergoes surgical correction in the first few days of life. Surgical options include end-to-end or end-to-side anastomosis. Surgery can be performed through traditional thoracotomy or video-assisted thoracostomy. Parenteral nutrition is provided for a few days until postoperative studies confirm that the anastomosis is intact and oral feedings can be given safely.
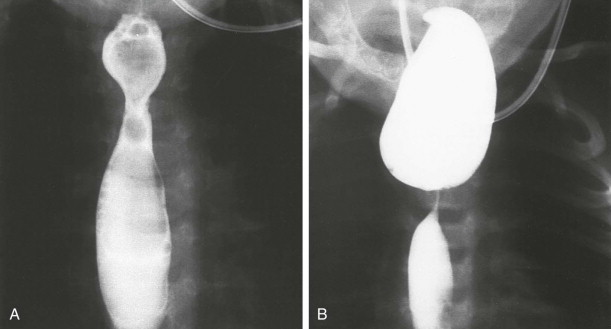
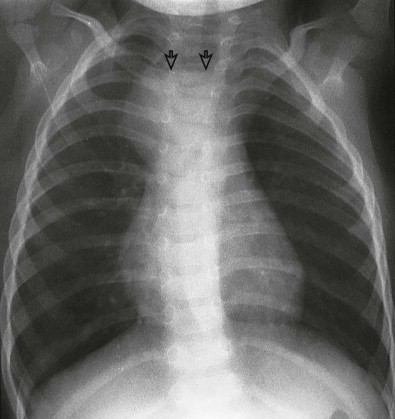
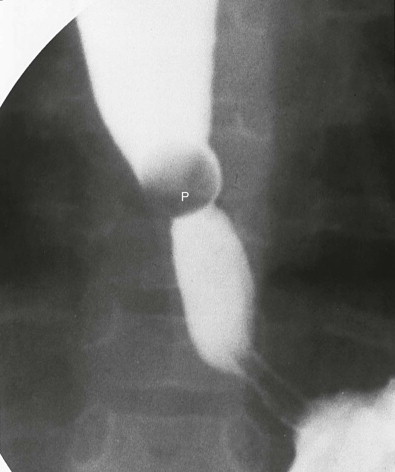
When there is a large gap between the proximal and distal segments, a common problem in the child with EA and no TEF, primary correction is usually not attempted in the neonatal period. Nutritional needs are met by a gastrostomy tube placed under surgical or radiologic guidance. The upper pouch is diverted so that secretions within it will not be aspirated. A neoesophagus is later created by interposing a segment of bowel or pulling the stomach into the esophagus, creating an anastomosis between the proximal esophagus and the stomach. This surgery is often delayed until the child is 1 year old. Nonsurgical anastomosis in EA may be created by magnetic lengthening of the esophageal segments with placement of a magnet in both ends of the upper and lower pouches, using the mouth and gastrostomy for catheter insertion. Postoperative complications are numerous, regardless of the type of repair performed. An anastomotic leak is seen in 10% to 20% of children who undergo primary repair in the neonatal period ( Fig. 114-18 ). Anastomotic narrowing or stricture formation ( Figs. 114-19 and 114-20 ) occurs in 15% to 30% of cases because of anastomotic leaks with scarring, ischemic change from tension on the anastomosis, or gastroesophageal reflux. A change in caliber at the anastomosis does not always indicate stricture but may instead result from residual dilation of the previously obstructed upper esophageal segment. Distal esophageal narrowing may be caused by congenital esophageal stenosis or gastroesophageal reflux ( Fig. 114-21 ). Bougienage and balloon dilation are used to treat these strictures.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


