Abstract
Neu-Laxova syndrome (NLS) is a rare autosomal recessive, lethal disorder. There are no formal diagnostic criteria for NLS. The most commonly described characteristics are intrauterine growth restriction, microcephaly, dysmorphic facies, central nervous system malformations, ichthyosis, and limb deformities. Studies have now suggested that the syndrome represents the severe end of the spectrum for serine deficiency disorders. Pregnancies at risk for Neu-Laxova or that have ultrasound findings consistent with the syndrome should be offered fetal diagnostic genetic testing. In pregnancies with Neu-Laxova that are continued, expectant management should be discussed with the parents to avoid unnecessary interventions, such as a cesarean section. Delivery usually occurs near term.
Keywords
Neu-Laxova syndrome, serine-deficiency disorders, microcephaly, limb deformities
Introduction
Neu-Laxova syndrome (NLS) is an autosomal recessive, lethal disorder. The syndrome was first described in 1971 by Neu et al. who reported three siblings who were either stillborn or died shortly after birth with microcephaly and multiple congenital anomalies. In 1972 Laxova et al. reported three siblings born to consanguineous parents with similar findings. In 1979 Lazjuk et al. described another case with similar findings and named the disorder. The prognosis for fetuses with NLS is poor because they either are delivered stillborn or die shortly after birth.
Disease
Definition
There are no formal diagnostic criteria for NLS. The diagnosis is based on clinical findings. The most commonly described characteristics are intrauterine growth restriction (IUGR), microcephaly, dysmorphic facies, central nervous system (CNS) malformations, ichthyosis, and limb deformities.
Prevalence and Epidemiology
NLS is a rare disorder with less than 100 reported cases. There does not appear to be a predilection for the syndrome in certain ethnic backgrounds.
Etiology and Pathophysiology
Until recently, the underlying pathogenesis and genetic etiology of NLS were unknown. Studies have now suggested that the syndrome represents the severe end of the spectrum for serine deficiency disorders. The serine biosynthesis pathway involves three enzymes: phosphoglycerate dehydrogenase, phosphoserine aminotransferase, and phosphoserine phosphatase. These three enzymes work together to convert 3-phospho-D-glycerate to L-serine. Individuals with defects in this pathway have been shown to have varying degrees of microcephaly, psychomotor retardation, severe spastic tetraplegia, nystagmus, intractable seizures, growth restriction, ichthyosis, cortical and subcortical hypotrophy, and abnormal myelination. Multiple studies have now reported cases of NLS with homozygous mutations in the genes responsible for the enzymes of the serine biosynthesis pathway; PHGDH (phosphoglycerate dehydrogenase), PSAT1 (phosphoserine aminotransferase), and PSPH (phosphoserine phosphatase). Although several NLS cases have been found to have a homozygous mutation in one of these three genes, there are still some cases in which no mutation has been found, suggesting the possible involvement of at least a fourth gene in the syndrome.
Manifestations of Disease
Clinical Presentation
The clinical presentation includes craniofacial and other abnormalities ( Table 134.1 ).
| CRANIOFACIAL |
|
| CENTRAL NERVOUS SYSTEM |
|
| LIMBS |
|
| OTHER |
|
Imaging Technique and Findings
Ultrasound.
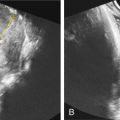
Prenatal ultrasound (US) diagnosis of NLS has been reported at 19 weeks’ gestation. The characteristic findings are severe IUGR, polyhydramnios, microcephaly with sloping forehead ( Fig. 134.1 ), exophthalmos, syndactyly, flexion deformities of the limbs, edema of hands and feet ( Fig. 134.2 ), and CNS abnormalities. Cataracts, cystic hygroma, clubbing of the feet, and hypodontia have also been reported.