Clinical Presentation
The patient is a 21-year-old man who has had neurologic symptoms for about 2 years. Initially, he had trouble holding a pen in the right hand. Over time, he has had more difficulty using his hands, especially the right hand. For the last 3 months, his symptoms have accelerated. He now has a spastic, unstable gait. The right leg is worse than the left. He has urinary hesitancy, but no incontinence. He has not experienced any falls. The patient has several café-au-lait spots and numerous subcutaneous soft tissue masses.
Imaging Presentation
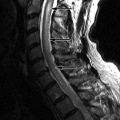
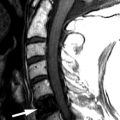
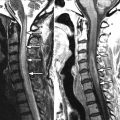
Sagittal and coronal T2-weighted images reveal numerous hyperintense foraminal and paraspinal soft tissue masses. On the sagittal images, the neural foramina are enlarged by the soft tissue masses. The findings are compatible with multiple neurofibromas in this patient with neurofibromatosis type 1 ( Fig. 51-1 ) .

Discussion
Neurofibromatosis (NF) is a subset of the phakomatoses. Phakomatoses, also known as neurocutaneous syndromes , consist of a group of disorders that tend to develop hamartomatous malformations and neoplastic growths affecting the skin, the nervous system, and other organs. The most common phakomatoses are NF, tuberous sclerosis, von Hippel-Lindau syndrome, and Sturge-Weber disease. NF is the most common of the phakomatoses. NF is classically divided into two types: NF type 1 (NF-1), also known as von Recklinghausen disease or peripheral neurofibromatosis, and NF type 2 (NF-2) or central neurofibromatosis.
NF-1 is much more common than NF-2, accounting for greater than 90% of all cases of neurofibromatosis. NF-1 occurs in 1 in 3500 live births. It is an autosomal dominant disorder in 50% to 60% of cases and is localized to chromosome 17 , which encodes for neurofibromin. Neurofibromin is a tumor suppressor gene. The remainder of the cases arise as spontaneous mutations. Diagnosic criteria that must be met for the diagnosis of NF-1 require the presence of two or more of the following: six or more café-au-lait spots, two or more neurofibromas, axillary or groin freckling, optic nerve glioma, two or more Lisch nodules (hamartomas of the iris), a distinctive bone lesion (such as hypoplasia of the sphenoid wing, pseudoarthrosis, severe kyphoscoliosis, or cortical thinning of a long bone), or a first-degree relative with NF-1. Some patients may have a very mild disease, whereas others are profoundly affected. Some of the complications occur at different times of life, which can delay the actual diagnosis of NF-1. Café-au-lait spots and external plexiform neurofibromas are seen within the first year of life and are present in 95% of all patients. Freckling and optic nerve gliomas are seen by 7 years of age. Cutaneous neurofibromas and Lisch nodules appear in teenage years or early adulthood. Malignancy and spinal plexiform neurofibromas are seen as adults.
The many spinal manifestations of NF-1 can be divided into bone and soft tissue abnormalities. Scoliosis is the most common musculoskeletal manifestation of NF-1 with an incidence of 71%. The scoliosis most often affects the thoracic spine. The most common scoliosis is a nondystrophic curve with a Cobb angle of less than 10 degrees and a kyphosis between 20 and 45 degrees. Dystrophic curves have a Cobb angle of greater than 10 degrees and a kyphosis of greater than 45 degrees. Primary bone involvement is known as mesodermal dysplasia . Mesodermal dysplasia causes the bone to be weaker and susceptible to erosion and remodeling, which can lead to scoliosis, secondary vertebral scalloping from tumoral compression, and lateral meningoceles. Soft tissue abnormalities include benign and malignant neurofibromas and schwannomas. Plexiform neurofibromas are present in 25% of cases of NF-1 and are considered pathognomonic for NF-1. A neurofibroma is a fusiform tumor mass that is located along the path of a peripheral nerve. Most plexiform neurofibromas are slow growing and benign; however, some neurofibromas may undergo malignant degeneration and invade and/or erode adjacent bones. Malignant degeneration occurs in 3% of all patients with NF-1 and is greatest in patients between the ages of 15 and 40.
There are no known predictive factors as to which tumors will undergo malignant degeneration. Most neurofibromas are asymptomatic. However, neurofibromas can affect large nerves, plexi, and spinal roots. The sacral plexus can be involved by tumor with compression of the ureters, rectum, and uterus. Plexiform neurofibromas involving spinal roots can cause pain from neural foraminal narrowing and cause spinal stenosis and/or cord compression/myelopathy depending on the level of abnormality. Other soft tissue involvement includes dural ectasias, which arise secondary to the pathologic (mesodermal dysplasia) bone with erosions of the vertebral bodies and the potential for formation of a lateral meningocele (pulsatile diverticula).
NF-2 is also an autosomal dominant disorder, which is located on chromosome 22 and encodes the 595-amino acid protein schwannomin (also known as merlin [moesin-ezin-radixin-like] protein), a tumor suppressor. Like NF-1, 50% of the reported cases of NF-2 are spontaneous mutations. However, NF-2 is much more rare than NF-1, seen in 1 of 40,000 people. There is no gender predilection. The term neurofibromatosis 2 is a misnomer because neurofibromas are not seen with NF-2.
The most common type of spinal nerve sheath tumors associated with NF-2 are schwannomas and are present in more than 80% of patients. In the spine of NF-2 patients, schwannomas and meningiomas have equal incidences and may occur simultaneously. Spinal schwannomas are usually small and asymptomatic, although in some patients they can be large and cause compression of the spinal cord or adjacent neural structures with resulting myelopathy and radicular pain. Peripheral schwannomas can arise from any nerve and can cause pain or impaired motor or sensory function. The diagnosis of NF-2 can be made on the basis of a patient having either bilateral vestibular nerve masses (nearly one half of all patients present with hearing loss), a positive family history with either a unilateral vestibular mass or any two of meningiomas, gliomas, schwannomas, and congenital cataracts. Because of the propensity of NF-2s for multiple intracranial and intraspinal neoplasms, NF-2 has also been called the MISME (multiple inherited schwannomas, meningiomas, and ependymomas) syndrome .
Comparing and contrasting NF-1 and NF-2, both entities are autosomal dominant. NF-1 is 10 times more common than NF-2. Both entities have intradural, extramedullary masses; however, these are seen more frequently with NF-2. Intramedullary masses are common with NF-2 and unusual with NF-1. The neurofibromas of NF-1 have malignant potential, whereas the schwannomas of NF-2 have no malignant potential. Both entities have a similar incidence of bone abnormalities; however, these are usually secondary abnormalities such as neural foraminal widening/scalloping secondary to tumors. Primary dysplastic bone changes are only seen with NF-1. Scoliosis also appears to be much more frequent with NF-1. Dural ectasias, although uncommon, are seen with NF-1 and not typical with NF-2.
Imaging Features
Because neurofibromatosis types 1 and 2 are syndromes, the imaging characteristics depend upon which finding(s) the patient has. The imaging discussion below pertains only to findings in the spine.
Neurofibromatosis-1
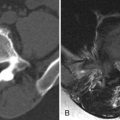
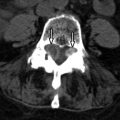
Plain radiographs can demonstrate the scalloping of the vertebra seen with NF-1 and other bone changes such as kyphosis/scoliosis and dysplastic (“ribbon”) or notched ribs ( Fig. 51-2 ) . The neurofibromas of NF-1 can be nodular or discrete or they can be plexiform neurofibromas that encase and enlarge the nerves. On computed tomography (CT), neurofibromas are hypodense with variable enhancement. There may be widening of the spinal canal and/or neural foramen secondary to dural ecstasia or the tumor itself ( Fig. 51-3 ) . On magnetic resonance imaging (MRI), neurofibromas are hypointense on T1-weighted sequences, hyperintense on T2-weighted sequences, and have a variable degree of enhancement ( Fig. 51-4 ) . Because neurofibromas and epidural/foraminal fat have similar signal intensity on T2-weighted sequences, it is best to perform fat-saturated, T2-weighted sequences. On T2 sequences imaged perpendicular to the axis, the neurofibroma appears like a target with a hyperintense rim and a low to intermediate signal intensity center ( Fig. 51-5 ) . The spinal neurofibromas often have a characteristic dumbbell shape with expansion of the neural foramen. There have been some studies that suggest that fluorodeoxyglucose positron emission tomography (FDG-PET) imaging may be useful in distinguishing benign from malignant neurofibromas. These studies suggest that malignant neurofibromas have higher standard uptake values (SUV) than benign ones. Patients with NF-1 frequently have dural ectasias and lateral meningoceles (cerebrospinal fluid [CSF]-filled widenings of the spinal canal with or without extension through the neural foramen). They have imaging characteristics similar to water; hypodense on CT, increased T1/decreased T2 signal on MR, and no evidence of enhancement ( Fig. 51-6 ) .