Congenital malformations encompass a diverse group of disorders that often present at birth, either as the result of genetic abnormalities, infection, errors of morphogenesis, or abnormalities in the intrauterine environment. Congenital disorders affecting the brain are now often diagnosed before delivery with the use of prenatal ultrasonography. Over the past several decades, there have been major advances in the understanding and management of these conditions. This review focuses on the most common cranial congenital malformations, limiting the discussion to the neurosurgically relevant aspects of arachnoid cysts, pineal cysts, Chiari malformations, and encephaloceles.
Congenital malformations encompass a diverse group of disorders that often present at birth, either as the result of genetic abnormalities, infection, errors of morphogenesis, or abnormalities in the intrauterine environment. Congenital disorders affecting the brain are now often diagnosed before delivery with the use of prenatal ultrasonography. Over the past several decades, there have been major advances in the understanding and management of these conditions. This article focuses on the most common cranial congenital malformations, limiting the discussion to the neurosurgically relevant aspects of arachnoid cysts, pineal cysts, Chiari malformations, and encephaloceles. In addition, cerebral arteriovenous malformations (AVMs) are included in this discussion, despite increasing evidence that they are not always the congenital and static lesions they were once thought to be.
Arachnoid cysts
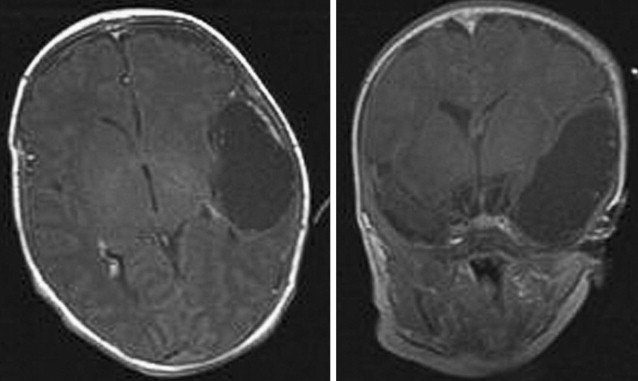
Arachnoid cysts are cerebrospinal fluid (CSF)-filled cavities surrounded by a wall of simple arachnoid cells. They develop within the arachnoid membrane as a result of membrane duplications or splitting. Although most arachnoid cysts are congenital, secondary arachnoid cysts result from CSF accumulations in patients after head injury, stroke, or central nervous system (CNS) infection. They should be differentiated from dermoid cysts; epidermoid cysts; neuroepithelial cysts; or leptomeningeal cysts occurring secondary to intracranial hemorrhage, infection, or trauma. They are typically asymptomatic and are discovered incidentally in children during evaluation for seizures, headaches, developmental delay, head trauma, or in infants with macrocrania ( Table 1 ). They occur in children at a prevalence rate of 0.5% to 2.6% with a male predominance ( Fig. 1 ). The location of arachnoid cysts has been well described. The most common location is the middle cranial fossa. Other common locations include retrocerebellar, supracerebellar, within the cerebellopontine angle, along the convexities, or in the quadrigeminal cistern ( Table 2 ). Middle fossa arachnoid cysts show a strong left-sided predominance for reasons that are not clear.
| Indication | Cases (%) |
|---|---|
| Concern for seizure | 16 |
| Headache | 14 |
| Cognitive dysfunction or developmental delay | 15 |
| Acute neurologic or mental status change | 13 |
| History of intracranial tumor | 11 |
| Trauma | 7 |
| History of hydrocephalus | 5 |
| Pituitary/endocrine issues | 3 |
| Intracranial hemorrhage | 2 |
| Other | 15 |
| Lesion Location | Total Number | Left | Right | Midline |
|---|---|---|---|---|
| Total | 309 | 139 | 84 | 86 |
| Anterior fossa | 6 | 1 | 5 | 0 |
| Middle fossa | 145 | 96 | 49 | 0 |
| Galassi type I | 99 | 64 | 35 | — |
| Galassi type II | 22 | 17 | 5 | — |
| Galassi type III | 24 | 15 | 9 | — |
| Posterior fossa | 118 | 35 | 25 | 58 |
| Posterior | 89 | 25 | 13 | 52 |
| Superior | 8 | 1 | 1 | 6 |
| Cerebellopontine angle | 20 | 9 | 11 | 0 |
| Convexity | 12 | 7 | 5 | 0 |
| Ventricular | 1 | 0 | 0 | 1 |
| Quadrigeminal plate | 18 | 0 | 0 | 14 |
| Sellar/suprasellar | 5 | 0 | 0 | 5 |
| Interhemispheric | 4 | 0 | 0 | 4 |
The etiology and embryology of arachnoid cysts are incompletely understood. Some think their association with other developmental abnormalities, such as autosomal dominant polycystic kidneys, heterotopias, neurofibromatosis type 1, and Marfan disease, suggests an underlying genetic cause. Given the common incidence of arachnoid cysts, however, it is probable that these associations are coincidental and may not carry any pathogenetic implications. Four histologic and ultrastructural differences have been noted between arachnoid cysts and the normal arachnoid membrane: hyperplastic arachnoid cells within the cyst wall, splitting of the arachnoid membrane at the margin of cyst wall, absence of traversing trabecular processes within the cyst, and a thick layer of collagen in the cyst wall. Arachnoid cysts are thought to represent developmental variants in the meninx primitiva, which surrounds the neural tube during differentiation of the mesenchyma. These CSF spaces, therefore, develop within the layers of the arachnoid membrane.
The indications for neurosurgical intervention in children with arachnoid cysts vary by institution and treating physician. Most cysts do not require surgical treatment. Indications for intervention in one single-institution review included progressive macrocephaly, headache, hydrocephalus, increased intracranial pressure, developmental delay, traumatic cyst rupture, behavioral concerns, and refractory epilepsy. Arachnoid cysts are common and great care must be taken when attempting to attribute nonfocal neurologic symptoms to an arachnoid cyst. Larger cysts and cysts within the anterior fossa and quadrigeminal plate were more likely to correlate with patient symptoms and receive recommendation for surgical treatment. Surgical treatments include craniotomy for cyst fenestration, endoscopic cyst fenestration, or CSF diversion with a cyst-to-peritoneum shunt.
The long-term behavior of arachnoid cysts is not well understood. Several case reports and small series have reported changes in cyst size, ranging from enlargement to spontaneous resolution. The natural history of arachnoid cysts was reviewed by Al-Holou and colleagues in a large single-institution review. Of 111 patients followed over a mean of 3.5 years, 11 arachnoid cysts increased in size, 13 decreased in size, and 87 remained stable. Younger age at presentation was associated with cyst enlargement and the need for neurosurgical intervention. In that series, no patient older than 4 years of age had cyst enlargement or developed new symptoms. The mechanism by which cysts enlarge is poorly understood; however, a number of theories have been proposed. The most widely accepted theory is that the arachnoid lining may function as a unidirectional ball valve, making CSF entry easier than CSF egress.
Pineal cysts
Pineal cysts are common incidental findings on brain imaging. They have been identified with increasing frequency since the advent of magnetic resonance (MR) imaging ( Fig. 2 ). Prior studies describing the prevalence of pineal cysts have estimated a population prevalence between 1.1% and 4.3%. A recent large single-institution series identified a 1.9% prevalence of pineal cysts on routine brain imaging in children and young adults, with a slightly increased prevalence in girls and older children.
On MR imaging, pineal cysts must be differentiated from cystic pineal region tumors, including teratomas, pineal parenchymal tumors, germ cell tumors, and low-grade astrocytomas, as well as arachnoid cysts in the adjacent quadrigeminal cistern. Although generally considered benign incidental findings, they have rarely been associated with neurologic sequelae, including hydrocephalus from aqueductal compression, Parinaud syndrome, and gaze paresis from tectal plate compression.
The etiology of pineal cysts is not known. The pineal gland is usually solid and less than 7 mm in diameter. When the pineal gland is greater than 1 cm in size, concern for the possibility of an underlying pineal mass increases. Pastel and colleagues reviewed the ultrastructure of benign pineal-region cysts to determine whether benign cysts have smooth walls or internal structure on MR imaging. The investigators found that the presence of thin walls supports the diagnosis of benign pineal cysts, and that internal septations or small internal cysts are common findings in benign cysts on MR imaging.
The clinical significance of these lesions was not well defined until recent analysis ( Table 3 ). Several series found that the majority of patients with pineal cysts had no change in cyst size on follow-up imaging and clinical evaluation. In one report of 32 patients with pineal cysts who underwent serial MR imaging, 75% of the cysts remained stable over time, 8% increased in size, and 16% regressed. In another study of 26 children, all incidentally found pineal lesions were stable in size after a maximum of 8 years of follow-up. These findings were supported by a recent single-institution study of 106 patients that were followed over a mean 3-year interval. In that report, 92% (98 of 106) of pineal cysts had no change in appearance on imaging (size or enhancement). Increased size at a mean patient age of 5.5 years was found in 6% (6 of 106), and 2% (2 of 106) had a change in imaging appearance without associated growth. Initial cyst size and imaging characteristics were not predictive of interval growth or change in pineal cyst size, and cysts with atypical features were not significantly more likely to change in size over time. Pineal cysts in younger patients were more likely to change than those found in older patients.
| Authors and Year | Follow-up Intervals | Number of Patients | Results | ||
|---|---|---|---|---|---|
| Increase in Cyst Size | Stable | Decrease in Cyst Size | |||
| Al-Holou et al, 2010 | Mean 3.0 y | 106 | 6 | 99 | 1 |
| Tamaki et al, 1989 | 3 mo – 4 y | 31 | 0 | 29 | 2 |
| Golzarian et al, 1993 | >1 y | 12 | 0 | 12 | 0 |
| Sawamura et al, 1995 | Median 1.5 y | 20 | 0 | 20 | 0 |
| Barboriak et al, 2001 | Mean 3.7 y | 32 | 3 | 24 | 5 |
| Totals | 201 | 9 | 184 | 8 | |
Pineal cysts almost never require surgical treatment. The indications for surgical intervention in several reported cases have included hydrocephalus, localizing neurologic symptoms, or the need to obtain a histologic confirmation for suspicious or rapidly expanding lesions. Management options for symptomatic pineal cysts have been a subject of debate over the past several years. Surgical options include open craniotomy for surgical excision followed by ventriculoperitoneal shunting for hydrocephalus, stereotactic cyst aspiration, and neuroendoscopy for lesion biopsy and endoscopic third ventriculostomy. In most cases, the existing literature does not support surgical treatment for these lesions, except in extraordinary cases.
Pineal cysts
Pineal cysts are common incidental findings on brain imaging. They have been identified with increasing frequency since the advent of magnetic resonance (MR) imaging ( Fig. 2 ). Prior studies describing the prevalence of pineal cysts have estimated a population prevalence between 1.1% and 4.3%. A recent large single-institution series identified a 1.9% prevalence of pineal cysts on routine brain imaging in children and young adults, with a slightly increased prevalence in girls and older children.
On MR imaging, pineal cysts must be differentiated from cystic pineal region tumors, including teratomas, pineal parenchymal tumors, germ cell tumors, and low-grade astrocytomas, as well as arachnoid cysts in the adjacent quadrigeminal cistern. Although generally considered benign incidental findings, they have rarely been associated with neurologic sequelae, including hydrocephalus from aqueductal compression, Parinaud syndrome, and gaze paresis from tectal plate compression.
The etiology of pineal cysts is not known. The pineal gland is usually solid and less than 7 mm in diameter. When the pineal gland is greater than 1 cm in size, concern for the possibility of an underlying pineal mass increases. Pastel and colleagues reviewed the ultrastructure of benign pineal-region cysts to determine whether benign cysts have smooth walls or internal structure on MR imaging. The investigators found that the presence of thin walls supports the diagnosis of benign pineal cysts, and that internal septations or small internal cysts are common findings in benign cysts on MR imaging.
The clinical significance of these lesions was not well defined until recent analysis ( Table 3 ). Several series found that the majority of patients with pineal cysts had no change in cyst size on follow-up imaging and clinical evaluation. In one report of 32 patients with pineal cysts who underwent serial MR imaging, 75% of the cysts remained stable over time, 8% increased in size, and 16% regressed. In another study of 26 children, all incidentally found pineal lesions were stable in size after a maximum of 8 years of follow-up. These findings were supported by a recent single-institution study of 106 patients that were followed over a mean 3-year interval. In that report, 92% (98 of 106) of pineal cysts had no change in appearance on imaging (size or enhancement). Increased size at a mean patient age of 5.5 years was found in 6% (6 of 106), and 2% (2 of 106) had a change in imaging appearance without associated growth. Initial cyst size and imaging characteristics were not predictive of interval growth or change in pineal cyst size, and cysts with atypical features were not significantly more likely to change in size over time. Pineal cysts in younger patients were more likely to change than those found in older patients.
| Authors and Year | Follow-up Intervals | Number of Patients | Results | ||
|---|---|---|---|---|---|
| Increase in Cyst Size | Stable | Decrease in Cyst Size | |||
| Al-Holou et al, 2010 | Mean 3.0 y | 106 | 6 | 99 | 1 |
| Tamaki et al, 1989 | 3 mo – 4 y | 31 | 0 | 29 | 2 |
| Golzarian et al, 1993 | >1 y | 12 | 0 | 12 | 0 |
| Sawamura et al, 1995 | Median 1.5 y | 20 | 0 | 20 | 0 |
| Barboriak et al, 2001 | Mean 3.7 y | 32 | 3 | 24 | 5 |
| Totals | 201 | 9 | 184 | 8 | |
Pineal cysts almost never require surgical treatment. The indications for surgical intervention in several reported cases have included hydrocephalus, localizing neurologic symptoms, or the need to obtain a histologic confirmation for suspicious or rapidly expanding lesions. Management options for symptomatic pineal cysts have been a subject of debate over the past several years. Surgical options include open craniotomy for surgical excision followed by ventriculoperitoneal shunting for hydrocephalus, stereotactic cyst aspiration, and neuroendoscopy for lesion biopsy and endoscopic third ventriculostomy. In most cases, the existing literature does not support surgical treatment for these lesions, except in extraordinary cases.
Pediatric arteriovenous malformations
Cerebral AVMs are abnormal connections between arteries and veins. Traditionally, AVMs have been considered congenital lesions, arising from a failure in embryogenesis of the cerebral vasculature. Some have postulated that AVMs occur during the third week of embryogenesis, during the formation and absorption of surface veins. Pediatric AVMs are thought to be dynamic in nature, constantly undergoing remodeling and angiogenesis. The role of abnormal angiogenesis via vascular endothelial growth factor (VEGF) in the formation and progression of AVMs is unclear. Studies have noted increased VEGF levels in children who have recurrence of their lesion after an initial resection as well as increased local expression of VEGF in AVM tissue. VEGF circulating in the plasma of patients with AVM has been found to be elevated compared with normal controls, with VEGF levels dropping soon after complete AVM resection.
Although AVMs are most often discovered during the third through fifth decades of life, they remain a common cause of spontaneous intracerebral hemorrhage (ICH) in children. Excluding hemorrhages of prematurity and early infancy, AVMs are the most frequently encountered structural cause of spontaneous ICH in childhood, and 80% to 85% of pediatric patients with AVM have intracranial hemorrhage as their presenting symptom. Other presenting symptoms in children include seizures, visual disturbance, headaches, and progressive neurologic dysfunction.
Three types of AVM have been identified. The most common type is high flow with a compact nidus, few arterial feeders, and draining veins. The diffuse variant with low flow and multiple arterial feeders and draining veins is less common. Recently, the linear vein-based morphology with multiple arterial feeders draining into a single vein has been described ( Figs. 3 and 4 ). Linear vein-based AVMs may be more prevalent in children than in adults.
The natural history of pediatric AVMs is not well understood. The annual intracranial hemorrhage rate and mortality from AVM rupture in children is higher than that for adults. Hemorrhage is associated with 25% mortality in children compared with the 6% to 10% annual mortality rate in adult patients. Factors that predict the bleeding risk of AVM hemorrhage are incompletely understood. Many reports suggest that children have a higher incidence of AVM hemorrhage compared with adults with an annual risk of 2%. AVM association with intranidal or pedicle aneurysms increases the risk of AVM rupture in children. Deep-venous drainage poses an increased hemorrhage risk. Location and a previous hemorrhage appear to be quite important in predicting hemorrhage risk, because deep-seated and cerebellar AVMs pose a greater risk. Size has been extensively studied with inconclusive results, because some find that smaller AVMs pose a greater risk of hemorrhage, whereas others suggest larger AVMs pose the greater hemorrhage risk.
AVMs in children are generally treated with the goal of eliminating risk of rehemorrhage. Current treatment options include microsurgical resection, stereotactic radiosurgery, and endovascular surgery. Surgical resection remains the gold standard for pediatric AVMs in accessible areas of the brain. The choice of optimal treatment, however, depends on the location and size of the AVM, as well as the age of patients. AVMs in areas of eloquent brain may be considered for treatment with radiosurgery or endovascular therapy, given the higher risk of postoperative neurologic deficits with open microsurgery. Microsurgery should be considered the treatment of choice in children with Spetzler-Martin grade I to III AVMs. The role of microsurgery in children with Spetzler-Martin grade IV and V AVMs is less clear. Ferch and Morgan retrospectively reviewed 46 adults treated surgically with grade IV or grade V AVMs, noting a 44% morbidity in those with a deep, perforating arterial supply and 10% morbidity in those without a deep component.
Endovascular surgery has an important role in the treatment of pediatric vascular conditions, including AVMs. Several groups have reported on children who underwent treatment of AVMs using a combination of preoperative embolization and microsurgery. In these series, all patients with Spetzler-Martin grade I to III AVMs had complete radiographic obliteration of their AVM with favorable clinical outcomes. The use of preoperative embolization has proven most beneficial in children with Spetzler-Martin grade II to V AVMs, thereby decreasing flow and size of the lesion, resulting in less blood loss during surgical resection.
Stereotactic radiosurgery has been used in children with AVMs in eloquent areas of the brain. Several groups have reviewed their obliteration rates by AVM volume, showing higher obliteration rates with smaller AVMs (<10 mL). The long-term effects of ionizing radiation on the developing brain are incompletely understood. Although 32% of children will show radiation-induced changes on MR imaging after treatment, the short-term neurologic complications in published series are estimated at 5% and include ataxia and cerebral edema.
Related posts:
 Demystifying Congenital Malformations for Imaging Specialists
Demystifying Congenital Malformations for Imaging Specialists
 Development and Dysgenesis of the Cerebral Cortex: Malformations of Cortical Development
Development and Dysgenesis of the Cerebral Cortex: Malformations of Cortical Development
 Imaging of Congenital Spine and Spinal Cord Malformations
Imaging of Congenital Spine and Spinal Cord Malformations
 Neurosurgical Management of Congenital Malformations and Inherited Disease of the Spine
Neurosurgical Management of Congenital Malformations and Inherited Disease of the Spine
 Congenital Malformations of the Temporal Bone
Congenital Malformations of the Temporal Bone
 Hemangiomas and Vascular Malformations of the Head and Neck: A Simplified Approach
Hemangiomas and Vascular Malformations of the Head and Neck: A Simplified Approach
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


