Abstract
Noonan syndrome is a disorder characterized by specific facial dysmorphisms, cardiovascular defects, short stature, and variable developmental delay. Noonan syndrome should be suspected prenatally in any fetus with cystic hygroma and normal karyotype, especially if a congenital heart defect is noted (particularly pulmonary stenosis). Because of the overlap between features of Noonan syndrome and other genetic disorders, postnatal physical examination in addition to molecular testing may be necessary for proper diagnosis. Noonan syndrome is genetically heterogeneous; it is mostly inherited in an autosomal dominant manner with complete penetrance and variable expressivity. Prenatal sonographic findings do not predict postnatal phenotype of Noonan syndrome. Life expectancy among patients with Noonan syndrome and minor cardiac anomalies is normal.
Keywords
cystic hygroma, Noonan syndrome, pulmonary stenosis
Introduction
Noonan syndrome (NS), first described by Dr. Jacqueline Noonan in 1968, is a disorder characterized by facial dysmorphisms, cardiovascular defects primarily consisting of pulmonary stenosis, short stature, and variable developmental delay. In addition, affected individuals often display pectus excavatum or carinatum, webbed neck, coagulopathies, and ocular anomalies. There is a great deal of variability in disease expression. NS is genetically heterogeneous; in almost all cases, it is autosomal dominant in nature, typically owing to a mutation in one of the known associated genes ( PTPN11, KRAS, SOS1, RAF1, BRAF, SHOC2, and NRAS). A rare form of NS is NS 2, which appears to be autosomal recessive in nature.
Disease
Definition
Hallmark features of NS include typical dysmorphisms (hypertelorism, downslanting palpebral fissures, and webbed neck), cardiovascular defects (in 80%), short stature, and pectus excavatum or carinatum. Other features include cognitive delay (30%), coagulopathies (50%), and other anatomic anomalies in varying degrees. In 61% of patients with NS, a mutation in an associated gene can be identified ( PTPN11 in 41% of cases, KRAS in 1.4%, SOS1 in 11.1%, RAF1 in 5%, BRAF in 1%, SHOC2 in 2%, and NRAS in 0.2% of cases).
Prevalence and Epidemiology
NS is estimated to be present in 1 : 1000 to 1 : 2500 individuals in the general population. The disorder affects all ethnic groups and occurs equally in males and females.
Etiology and Pathophysiology
Several genes have been identified as causes of NS; nevertheless, the pathophysiology of this condition is not well delineated. Each gene ( PTPN11, KRAS, SOS1, RAF1, BRAF , SHOC2 , and NRAS ) is part of the RAS/RAF/MEK/ERK signal transduction pathway, which is important in cell growth regulation. Somatic mutations of several of these genes have been identified in tumor cells; however, it remains unclear how germline mutations in these genes lead to the wide array of anomalies found in individuals with NS.
Manifestations of Disease
Clinical Presentation
NS shares many features with Turner syndrome, and it is important that normal karyotyping be confirmed first in a female in whom the diagnosis of NS is suspected. In addition, the NS phenotype overlaps with many other disorders, including LEOPARD syndrome (which is allelic to NS caused by either PTPN11 or RAF1 mutations), cardiofaciocutaneous syndrome (which can also rarely be caused by KRAS mutations), neurofibromatosis–NS, and Costello syndrome. Molecular testing may help delineate which of these diagnoses is correct in an individual with features consistent with more than one of these syndromes. There are no set diagnostic criteria for NS; however, the diagnosis can be made in an individual in whom several of the following cardinal features are noted including, but not limited to :
- •
postnatal growth failure
- •
mild cognitive delay
- •
cardiovascular defects
- •
pulmonary stenosis
- •
coarctation of aorta ( Fig. 135.1 )

Fig. 135.1
Long-axis view of the left ventricle in a fetus with coarctation of the aorta shows a narrowed aortic root.
- •
hypertrophic obstructive cardiomyopathy ( Fig. 135.2 )

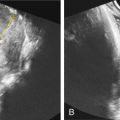
Fig. 135.2
(A) pleural effusion and (B) facial dysmorphism in a fetus with postnatal confirmation of NS.
License Number: 3854930403226.
- •
abnormal electrocardiogram
- •
- •
skeletal
- •
pectus excavatum or carinatum and/or cubitus valgus
- •
fifth finger clinodactyly and/or brachydactyly
- •
scoliosis
- •
- •
facial dysmorphisms (see Fig. 135.2 )
- •
hypertelorism and ptosis with downslanting palpebral fissures
- •
epicanthal folds
- •
triangular facies
- •
low, posteriorly rotated, fleshy ears
- •
deeply grooved philtrum, upper lip vermilion border with high peaks
- •
micrognathia and other oral manifestations
- •
- •
neck abnormalities including: webbed neck or cystic hygroma (see Fig. 135.2 ), broad neck with low posterior hairline
- •
widely spaced nipples
- •
prenatal and postnatal lymphedema
- •
genitourinary (renal anomalies, cryptorchidism in males, and incontinence)
- •
ocular findings (myopia, blue-green irides)
- •
ear–nose–throat involvement
- •
eustachian tube dysfunction including potential hearing loss
- •
speech delay
- •
- •
woolly hair
- •
keratoses
- •
coagulopathies
- •
myeloproliferative disorder
- •
malignant schwannoma
- •
chronic pain
- •
psychiatric disease
Imaging Technique and Findings
Ultrasound.
First-trimester ultrasound (US) performed between 10 weeks and 14 weeks may show evidence of cystic hygroma ( Fig. 135.3 ) or increased nuchal translucency in the presence of normal fetal karyotype. Later in gestation, US may reveal a thickened nuchal fold, polyhydramnios, ascites, pleural effusion (see Fig. 135.2 ), macrocephaly, shortened long bones, cardiac and/or renal malformations. A recent review found 75% of infants with NS had one US abnormality, whereas 25% had two or more abnormal sonographic findings. However, the sonographic findings are often nonspecific and do not predict postnatal phenotype of NS.