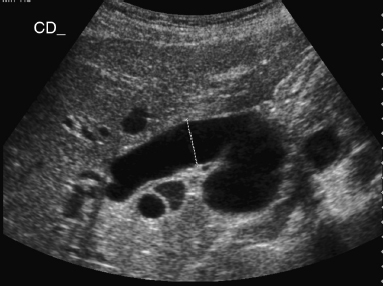
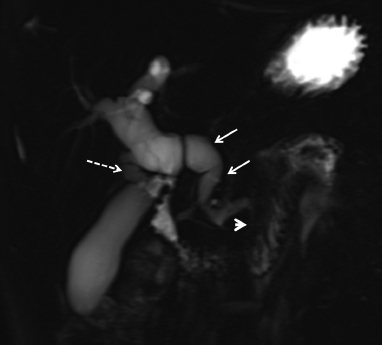
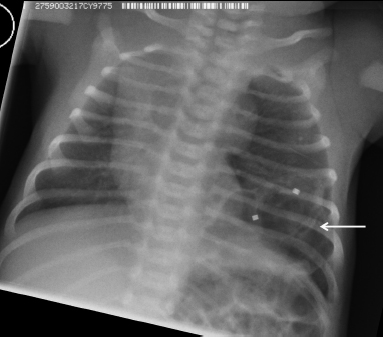
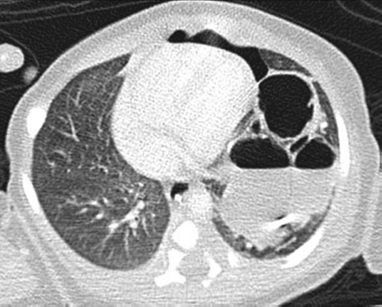
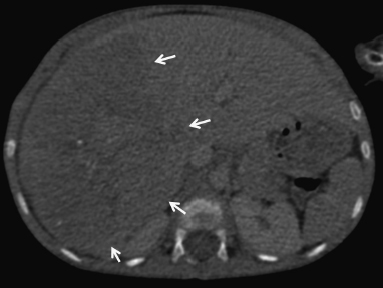
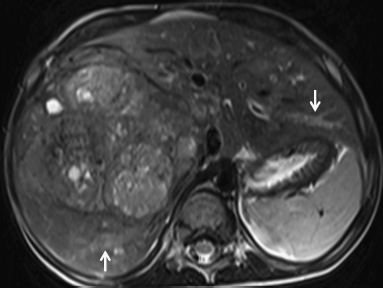
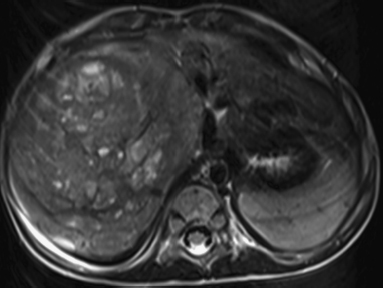
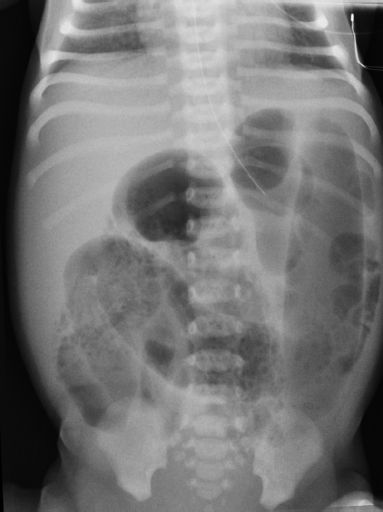
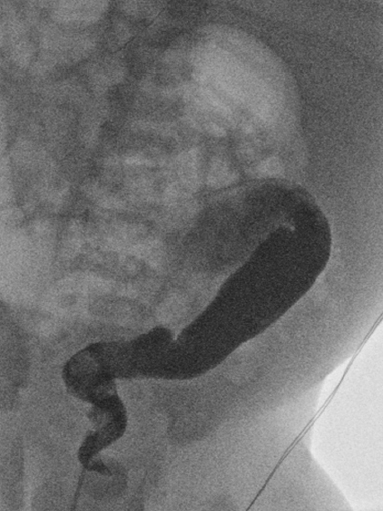
6 Introduction to Paediatric Imaging Paediatric imaging is part of the core radiology training curriculum1; therefore, it should come as no surprise to find a significant number of cases appearing in postgraduate radiology examinations. From our collective experience, some examiners may have predominantly paediatric films in the viva, so be prepared. After clearing the hurdle of the examination, every radiologist is likely to encounter paediatric imaging regardless of their chosen radiological subspecialty. Furthermore, as paediatric imaging includes every organ system, a good grasp of both normal variants and common pathology will prove very useful for the future. It is important to be aware of the major differences between paediatric and adult imaging. In children, ultrasound, plain radiographs, and fluoroscopy form the foundation of imaging. Plain radiographs are the cornerstone in the assessment of chest disease for children of all ages. Fluoroscopy is used extensively for abnormalities of the gastrointestinal and genitourinary tracts. Ultrasound is a very useful tool, especially for abdominal abnormalities—children normally have little body fat, allowing excellent visualisation of the solid organs. For a child presenting with abdominal pain or distension, ultrasound is the imaging modality of choice. Within the chest, ultrasound can detect pleural effusions and empyemas; it can also act as a problem-solving tool if there is, for example, concern on the plain radiograph regarding thymus versus an anterior mediastinal mass—thymic tissue has a distinctive appearance on ultrasound. Children are more susceptible to the detrimental effects of radiation than adults, so every effort should be made to reduce the dose and apply the “as low as reasonably achievable” principle. There should be strict justification for performing studies involving radiation. However, if performed, paediatric-specific protocols should be used; this is especially true for CT and fluoroscopy. Other considerations include gonad, thyroid, and lens shielding, suitable collimation, added filtration, and grid removal. Movement artefact can ruin image quality, but this can be diminished with the use of short exposure times, immobilisation, sedation, or distraction techniques to reduce patient motion.2 CT and MRI are primarily used to problem-solve, with MRI being increasingly used more than CT, especially now high-resolution and isotropic sequences can give excellent anatomical detail. MRI is ideal for staging abdominal tumours and also for non-oncological pathology, such as congenital anomalies involving the genitourinary tract. Nonaccidental injury must be on the examination candidate’s awareness at all times. Always check the bones on every plain film for fractures; for example, fractures involving the ribs, scapulae, and sternum on a chest radiograph are more suspicious for nonaccidental injury. If nonaccidental injury is suspected, the appropriate paediatric clinician needs to be alerted immediately, and this must be documented in the report. Following discussion, a full skeletal survey may be performed; for standard views, see the report from the Royal College of Radiologist of the United Kingdom.3 There must be specific considerations for paediatric images in the examination: • Capitellum—8 months • Radial head—3–4 years • Internal (medial) epicondyle—5 years • Trochlea—7 years • Lateral epicondyle—11 years. 1. Royal College of Radiologists. Specialty Training Curriculum for Clinical Radiology. May 2010. http://www.rcr.ac.uk/content.aspx?PageID=1805 (accessed June 2012) 2. Raissaki MT. Pediatric radiation protection. Eur Radiol 2004;14:74–83 3. Royal College of Radiologists and Royal College of Paediatrics and Child Health. Standards for Radiological Investigations of Suspected Non-Accidental Injury – March. 2008. Intercollegiate Report from the Royal College of Radiologists and Royal College of Paediatrics and Child Health. http://www.rcr.ac.uk/docs/radiology/pdf/RCPCH_RCR_final.pdf (accessed June 2012) A 2-month-old infant presents with jaundice (Fig. 6.1.1). This is a selected ultrasound image of the liver. There is a fusiform dilated structure in the region of the common hepatic duct, with no debris present (Fig. 6.1.1, between cursors). I would like to review further images to assess the remaining biliary system and gallbladder. This appearance is suggestive of a choledochal cyst, and I would recommend magnetic resonance cholangiopancreatography (MRCP) for further evaluation. This is the MRCP image (Fig. 6.1.2). This is a maximum-intensity projection image from an MRCP examination. There is fusiform dilatation of the common hepatic and common bile ducts (Fig. 6.1.2, arrows). There is an abrupt calibre change in the common bile duct just before the junction with the pancreatic duct (Fig. 6.1.2, arrowhead). The left and right hepatic ducts are also dilated. The gall-bladder appears normal. The cystic duct originates from the abnormal dilated common duct (Fig. 6.1.2, dashed arrow). I cannot see any filling defects within the biliary tree. The pancreatic duct is not dilated. I would assess the other sequences for evidence of pancreatitis. The appearance of the common hepatic and the common bile duct are those of a choledochal cyst. I would inform the referring team. Ultrasound is the first-line investigation for jaundice in this group of patients, with even subtle changes in the calibre of the common or hepatic bile ducts warranting further investigation, as complications are substantial in the long term. • Calculus formation. Ultrasound may depict echogenic stones with posterior acoustic shadowing in the choledochal cyst or biliary system. These will also be seen as filling defects on the MRCP. • There may be pancreatitis. • Cholangitis and hepatic abscess formation may be seen. • Choledochal cyst rupture. This may result in a collection or free fluid around the liver. • Biliary cirrhosis. Imaging features depend on the stage of the disease. If there is a late presentation, there will be an irregular, shrunken liver with signs of portal hypertension, ascites, and splenomegaly. • Cholangiocarcinoma. This may be subtle on imaging, but look for a mass or abnormal enhancement as a cause for focal intrahepatic duct dilatation. There is no differential diagnosis for this appearance: any obstruction of the bile ducts will result in global dilatation of the bile ducts to the point of obstruction. Occasionally, on the ultrasound examination, other cystic structures may cause confusion. • MRCP should not demonstrate any communication with the biliary system with no biliary dilatation. • No uptake of radiotracer will be seen on the technetium 99m DISIDA scan. Choledochal cysts are an uncommon entity. They usually present early in life, and there is a classical triad of jaundice, an abdominal mass, and abdominal pain. • Type I: the most common variety (80 to 90%), involving saccular or fusiform dilatation of a portion of or the entire common bile duct (CBD), with normal intrahepatic ducts. • Type II: a true isolated diverticulum protruding from the CBD. • Type III: a choledochocoele arising from dilatation of the duodenal portion of the CBD or where the pancreatic duct meets it. • Type IV: characterised by multiple dilatations of the intrahepatic and extrahepatic biliary tree (IVa) or only the extrahepatic bile ducts (IVb). • Type V: Caroli’s disease, with cystic dilatation of the intrahepatic biliary ducts. Rozel C, Garel L, Rypens F, et al. Imaging of biliary disorders in children. Pediatr Radiol 2011;41(2):208–220 Kim OH, Chung HJ, Choi BG. Imaging of the choledochal cyst. Radiographics 1995;15(1):69–88 Chavhan GB, Babyn PS, Manson D, Vidarsson L. Pediatric MR cholangiopancreatography: principles, technique, and clinical applications. Radiographics 2008;28(7):1951–1962 A neonate presents with respiratory distress (Fig. 6.2.1). This is a frontal chest radiograph of a neonate, with a prominent thymic shadow noted. There is an area of increased opacification with multiple lucencies occupying the lower aspect of the left lung (Fig. 6.2.1, arrow), which have the appearance of thin-walled cysts. The mediastinum is displaced to the right. The right lung appears normal. The abnormal area of left-sided opacification does not erode or scallop the posterior ribs. Two small metallic densities are seen in the left lower zone. Normal bowel gas is seen beneath the left diaphragm. These features are most likely to represent a congenital cystic adenomatoid malformation (CCAM). I would discuss these findings with the referring team and confirm the diagnosis with a chest CT. This is a chest CT image from the same patient (Fig. 6.2.2). This is an axial image on lung windows. There is a large multicystic mass centred on the left lower lobe, with air–fluid levels, raising the possibility of a communication with the bronchial tree. This is causing a mediastinal shift to the right, as seen on the radiograph. There are no anomalous vessels associated with the mass, and the diaphragm appears intact. The CT features confirm the diagnosis of CCAM. When assessing lesions on CT in the neonate, you must check the following: • Sequestration does not usually contain gas, unless there is concomitant infection. • Assessment of feeding and draining vessels is crucial to the diagnosis. • On plain radiographs, initially opacified areas may become progressively lucent on follow-up films. • The diagnosis is confirmed on CT imaging, which shows overexpansion of a single or multiple lung lobes, most frequently the left upper lobe. • Look for lucencies within the chest that resemble bowel and are adjacent to the diaphragm. • There may be reduced bowel gas within the abdomen and low volumes in the chest. • The position of the nasogastric tube and intravenous lines may provide clues. • This shows bubble-like or linear small lucencies within the lung that are classically uniform in size, unlike in CCAM. • On CT, a central arteriole is seen within each cystic area, compared with the empty cysts seen in CCAM. CCAM is a congenital lung mass arising from disorganised hamartomas and adenomatoid proliferation of the primary bronchioles, which are in continuity with the bronchial tree. The majority are located within a single lobe and are rarely seen in the right middle lobe. There are three types of CCAM: Type II and type III CCAMs are commonly associated with other congenital anomalies, such as cardiac abnormalities, renal agenesis, and jejunal atresia. Lee EY, Boiselle PM, Cleveland RH. Multidetector CT evaluation of congenital lung anomalies. Radiology 2008;247(3): 632–648 Griffin N, Devaraj A, Goldstraw P, Bush A, Nicholson AG, Padley S. CT and histopathological correlation of congenital cystic pulmonary lesions: a common pathogenesis? Clin Radiol 2008;63(9):995–1005 A neonate presents with severe respiratory distress from birth (Fig. 6.3.1). This is a plain chest and abdominal radiograph of a neonate. The endotracheal tube is positioned low, and may be withdrawn slightly. The distal aspect of the nasogastric tube is at T2, and is incorrectly positioned. The tip of the left femoral line is deviated to the right of the midline. Multiple circular lucent areas with discernible walls fill the left hemithorax, extending into the left upper quadrant of the abdomen. The left hemidiaphragm is not visible. A larger lucent area is seen overlying the midline. There is marked mediastinal displacement to the right. Gas-filled loops of bowel are seen within the abdomen. No free intraperitoneal gas is evident. No bony abnormalities are seen. The most likely diagnosis is left-sided congenital diaphragmatic hernia (CDH), with bowel loops in the left hemithorax and possibly stomach overlying the mediastinum. I would recommend urgent referral to the paediatric surgical team. • The nasogastric tube may be curled within an intrathoracic stomach. • The tip of the umbilical venous catheter may be located within the left hemithorax, indicating that the liver has herniated into the left side of the chest; this is an uncommon but important observation to make as it is associated with a worse prognosis. • The umbilical arterial catheter passes from the umbilical artery into the internal iliac artery (where it is seen to “loop” on the plain film), and then into the aorta. On the plain film, the tip should be sited to the left of the midline between T6 and T10 or below L2, away from the major aortic branches. In the United Kingdom, it is usually positioned in the more cranial position. • The umbilical venous catheter passes from the umbilical vein to the left portal vein, ductus venosus, and finally the inferior vena cava. The tip should lie in the region of the cardiophrenic recess, that is, within the cephalad portion of the inferior vena cava. Congenital cystic adenomatoid malformation (CCAM): • Cardiac anomalies are reported in up to 50% and include ventricular septal defects, transposition of the great vessels, and tetralogy of Fallot. • Oesophageal atresia may occur, with absence of bowel gas and opacification of the affected chest. Taylor GA, Atalabi OM, Estroff JA. Imaging of congenital diaphragmatic hernias. Pediatr Radiol 2009;39(1):1–16 Deprest J, Nicolaides K, Done’ E, et al. Technical aspects of fetal endoscopic tracheal occlusion for congenital diaphragmatic hernia. J Pediatr Surg 2011;46(1):22–32 A 3-week-old neonate presents with dyspnoea (Fig. 6.4.1). This is a frontal chest radiograph of a neonate. There is a hyperlucent area with lung markings filling most of the left hemithorax. This is causing mediastinal shift to the right with increased opacification in most of the right lung as well as the left base, which may be due to collapse. The most likely differential diagnosis is congenital lobar emphysema (CLE), but other differentials include congenital cystic adenomatoid malformation (CCAM). I would like to check any previous imaging if available, and I would confirm the diagnosis with CT. These are selected images from the CT chest examination (Figs. 6.4.2 and 6.4.3). These are coronal and axial images on lung window settings through the chest. There is overexpansion of the left upper lobe, resulting in atelectasis of the left lower lobe and a portion of the right lung. I would like to assess the remainder of the study for bronchial atresia. If the bronchial tree is normal, the most likely diagnosis is CLE. I would refer the patient to the paediatric respiratory unit for further management. When dealing with hyperlucent lung on a plain film, assess the following: • Congenital lobar emphysema in a single lobe may result in collapse and opacification of the ipsilateral lobe and contralateral lung. • Volume loss of a single lobe causes opacification of that lobe, but lucency in the adjacent ipsilateral lobes and the contralateral lung. • Volume loss of an entire lung (pulmonary agenesis or hypoplasia) causes opacification of that lung as well as lucency of the contralateral lung. • Assess the bronchial tree and size of pulmonary vessels: • The presence of cysts within the hyperlucent area may be due to CCAM or congenital diaphragmatic hernia (CDH). Look for varying size of the cysts in CCAM, and check for continuation of bowel loops below the level of the diaphragm in CDH. • CT may differentiate CLE (bronchovascular bundles at the periphery of lucent areas) from persistent pulmonary interstitial emphysema (bronchovascular bundles at the centre of cysts). • The thoracic wall may be underdeveloped, with loss of volume on the affected side. • This is associated with cardiac abnormalities, scimitar syndrome, diaphragmatic abnormalities, gastrointestinal and genitourinary abnormalities (cystic renal disease), tracheooesophageal fistula, and imperforate anus. • Bronchial mucous plugs can be visualised on CT imaging. • Appearances typically resolve on follow-up imaging. • The characteristic CT finding is a central opacity that may have a tubular shape or may contain an air–fluid level representing the obstructed, dilated, and mucoid-filled segmental or subsegmental bronchus. • Hyperinflation due to collateral air drift and a relative dearth of vessels due to obstructed or nonexistent blood supply in the involved segment of lung are also frequently seen on CT imaging. Lee EY, Boiselle PM, Cleveland RH. Multidetector CT evaluation of congenital lung anomalies. Radiology 2008;247(3):632–648 A 2-year-old child presents with increasing abdominal girth (Fig. 6.5.1). This is a noncontrast axial CT image through the upper abdomen of a young child. There is a large predominantly low-attenuation lesion in the right lobe of the liver (Fig. 6.5.1, arrows). This contains a few small foci of high density, likely to be calcification. No biliary dilatation or ascites is seen. I would like to review the remainder of the study and ask for contrast-enhanced images. Any further evaluation of this abnormality should be performed with MRI. These are selected images from an MRI examination of the same patient (Figs. 6.5.2 and 6.5.3). These are selected axial T2-weighted MRI scans through the upper abdomen. In the right lobe of the liver, there is a large multilobulated lesion that is well circumscribed and demonstrates mixed signal intensity. I cannot see any biliary duct dilatation. There is a mass effect with displacement of the inferior vena cava to the left of midline. I cannot see the right hepatic or portal vein, but I am able to identify the left and right hepatic veins (Fig. 6.5.2, arrows). I would like to review all the images to assess for vascular occlusion and the presence of a separate intra-abdominal mass, and to confirm that this is a solitary liver lesion. The findings in a child of this age are likely to represent a primary hepatic tumour, such as a hepatoblastoma. I would recommend referral to a specialist paediatric oncology centre and correlation with serum alphafetoprotein levels. The assessment of a liver mass may be difficult due to the lack of pathognomonic imaging characteristics. However, careful identification of following features will narrow the differential diagnosis: • Always check the patency of the portal and hepatic veins, hepatic artery, inferior vena cava, and aorta with colour Doppler imaging. • Look carefully for other subtle hepatic lesions. • Always assess for biliary dilatation as decompression may be necessary depending on the patient’s clinical status. • Beckwith–Weidemann syndrome. There is likely to be hepatomegaly, in addition to hypertrophy of other solid abdominal and pelvic viscera. • Familial adenomatosis polyposis coli. Look at the bowel for large polyps. Das CJ, Dhingra S, Gupta AK, Iyer V, Agarwala S. Imaging of paediatric liver tumours with pathological correlation. Clin Radiol 2009;64(10):1015–1025 A neonate that has failed to pass meconium (Fig. 6.6.1). This is a supine abdominal radiograph of a neonate. There are multiple, markedly dilated loops of bowel within the abdomen with a paucity of bowel gas seen in the pelvis. A nasogastric tube is correctly positioned. There is no free gas. The appearances are those of a distal bowel obstruction. The differential diagnosis includes Hirschsprung’s disease and meconium plug syndrome. If the neonate is not septic, I would perform a water-soluble contrast enema to assess the extent and cause of obstruction. This is an image from the contrast examination (Fig. 6.6.2). This is a selected anteroposterior image from a contrast enema study. The sigmoid and descending colon are dilated, and there is a transition point in the region of the rectosigmoid junction. The rectum is abnormally narrowed. I would like to view the rest of the series. The most likely diagnosis is Hirschsprung’s disease, and I would inform the paediatric surgeons. When shown a contrast enema, comment on the following: • Hirschsprung’s disease may involve the entire colon, producing a “microcolon” appearance. • Other causes of microcolon include meconium ileus and ileal atresia. • Meconium plug syndrome demonstrates a normal rectosigmoid ratio with a small left colon and transition point at the splenic flexure. This may be difficult to differentiate from a long segment of Hirschsprung’s disease. • A contrast enema should demonstrate intraluminal colonic filling defect (meconium plugs).
 State the imaging modality and body part.
State the imaging modality and body part.
 Knowing the age of the patient can help narrow your list of differentials: neonate (less than 28 days old), infant (less than 1 year old), and then child can be used up to the age of 16 years. Look at the bones for clues to the age. For example, the proximal femoral epiphysis begins to ossify after 3 months of age, and the humeral head is not ossified in premature neonates.
Knowing the age of the patient can help narrow your list of differentials: neonate (less than 28 days old), infant (less than 1 year old), and then child can be used up to the age of 16 years. Look at the bones for clues to the age. For example, the proximal femoral epiphysis begins to ossify after 3 months of age, and the humeral head is not ossified in premature neonates.
 The use of the various ossification centres of the elbow are important in the assessment of fractures, but can also help with ages:
The use of the various ossification centres of the elbow are important in the assessment of fractures, but can also help with ages:
 Be aware of associated findings and satisfaction of search. For example, if you do not realise that tracheo-oesophageal fistula is associated with the VACTERL syndrome, you will probably miss the vertebral abnormalities and you will definitely not ask for further renal imaging to get you those extra points.
Be aware of associated findings and satisfaction of search. For example, if you do not realise that tracheo-oesophageal fistula is associated with the VACTERL syndrome, you will probably miss the vertebral abnormalities and you will definitely not ask for further renal imaging to get you those extra points.
 Always ask for old films and review all of the available imaging.
Always ask for old films and review all of the available imaging.
 Listen to the examiner: the clinical history is important and could be the deciding factor in determining your differential diagnosis.
Listen to the examiner: the clinical history is important and could be the deciding factor in determining your differential diagnosis.
References
6.1 Choledochal Cyst
Clinical History
Ideal Summary
Examination Tips
 Recommend MRI rather than CT as the next line of investigation in this age group.
Recommend MRI rather than CT as the next line of investigation in this age group.
 Check for any complications of a choledochal cyst:
Check for any complications of a choledochal cyst:
Differential Diagnosis
 Hepatic cyst:
Hepatic cyst:
Notes
 Classification is based on site of the cyst or dilatation:
Classification is based on site of the cyst or dilatation:
 In cases where the diagnosis is unclear, radionuclide studies will show tracer uptake into the choledochal cyst.
In cases where the diagnosis is unclear, radionuclide studies will show tracer uptake into the choledochal cyst.
 Treatment depends on the type of choledochal cyst, and ranges from excision and biliary reconstruction to hepatic lobectomy (when there is stricture or stenosis).
Treatment depends on the type of choledochal cyst, and ranges from excision and biliary reconstruction to hepatic lobectomy (when there is stricture or stenosis).
 Biliary atresia is associated with choledochal cysts in neonates. Hepatobiliary scintigraphy with technetium 99m DISIDA scanning can be performed; lack of excretion into the small bowel is suggestive of biliary atresia.
Biliary atresia is associated with choledochal cysts in neonates. Hepatobiliary scintigraphy with technetium 99m DISIDA scanning can be performed; lack of excretion into the small bowel is suggestive of biliary atresia.
Bibliography
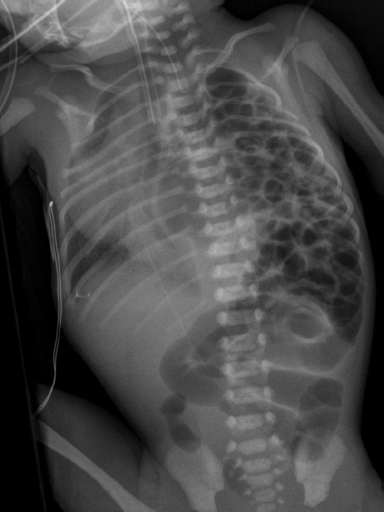
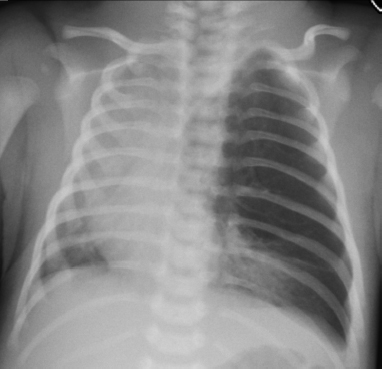
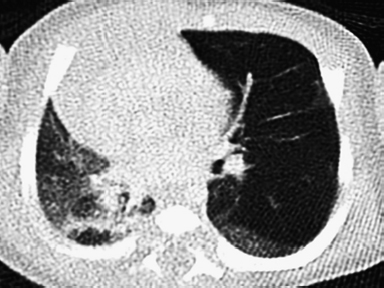
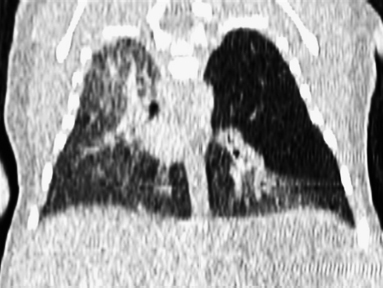
6.2 Congenital Cystic Adenomatoid Malformation
Clinical History
Ideal Summary
Examination Tips
 Evaluate each lesion using both lung parenchymal and mediastinal window settings. If these are not initially available, the candidate will be expected to ask for them.
Evaluate each lesion using both lung parenchymal and mediastinal window settings. If these are not initially available, the candidate will be expected to ask for them.
 Comment on the pattern of the abnormality: Is it soft tissue, cystic, nodular, or hyperexpanded lung parenchyma?
Comment on the pattern of the abnormality: Is it soft tissue, cystic, nodular, or hyperexpanded lung parenchyma?
 Assess the distribution of abnormality: Is it confined to a particular lobe? Is it diffuse? Does it have an upper or lower zone predilection? Congenital cystic adenomatoid malformation is typically isolated to a single lobe with no lobar predominance.
Assess the distribution of abnormality: Is it confined to a particular lobe? Is it diffuse? Does it have an upper or lower zone predilection? Congenital cystic adenomatoid malformation is typically isolated to a single lobe with no lobar predominance.
 If the mass is responsible for significant mediastinal shift, comment upon compression of the bronchi, subsequent atelectasis, and even compression of the large vessels, as this assumes importance in a child.
If the mass is responsible for significant mediastinal shift, comment upon compression of the bronchi, subsequent atelectasis, and even compression of the large vessels, as this assumes importance in a child.
 Carefully evaluate the tracheobronchial tree for areas of narrowing that could represent tracheomalacia. Beware of the possibility of bronchial atresia.
Carefully evaluate the tracheobronchial tree for areas of narrowing that could represent tracheomalacia. Beware of the possibility of bronchial atresia.
 Evaluate the blood supply on mediastinal window settings: an arterial feeding vessel from the thoracic aorta and draining veins into the inferior pulmonary veins or inferior vena cava is seen in sequestration and should prompt the diagnosis.
Evaluate the blood supply on mediastinal window settings: an arterial feeding vessel from the thoracic aorta and draining veins into the inferior pulmonary veins or inferior vena cava is seen in sequestration and should prompt the diagnosis.
 If the lesion erodes or scallops the posterior ribs, think of a thoracic neuroblastoma.
If the lesion erodes or scallops the posterior ribs, think of a thoracic neuroblastoma.
Differential Diagnosis
 Pulmonary sequestration:
Pulmonary sequestration:
 Congenital lobar emphysema:
Congenital lobar emphysema:
 Congenital diaphragmatic hernia:
Congenital diaphragmatic hernia:
 Pulmonary interstitial emphysema:
Pulmonary interstitial emphysema:
Notes
 Type I (50%): single or multiple large cysts
Type I (50%): single or multiple large cysts
 Type II (40%): multiple small cysts
Type II (40%): multiple small cysts
 Type III (10%): solid with microscopic cysts
Type III (10%): solid with microscopic cysts
Bibliography
6.3 Congenital Diaphragmatic Hernia
Clinical History
Ideal Summary
Examination Tips
 The plain film appearances depend on the contents of the hernia: stomach and bowel are gasfilled, while liver is radiopaque.
The plain film appearances depend on the contents of the hernia: stomach and bowel are gasfilled, while liver is radiopaque.
 On the first film, before gas has entered the gastrointestinal tract, the affected hemithorax may be of increased opacification. On subsequent films, gas is usually seen within the bowel loops.
On the first film, before gas has entered the gastrointestinal tract, the affected hemithorax may be of increased opacification. On subsequent films, gas is usually seen within the bowel loops.
 The position of lines and tubes should be carefully assessed and commented upon as this may provide clues to the diagnosis:
The position of lines and tubes should be carefully assessed and commented upon as this may provide clues to the diagnosis:
 Ultrasound can confirm loops of bowel, stomach, or solid viscera within the chest with CDH.
Ultrasound can confirm loops of bowel, stomach, or solid viscera within the chest with CDH.
 If there is a small CDH, ultrasound may be able to depict the hemidiaphragm and the defect.
If there is a small CDH, ultrasound may be able to depict the hemidiaphragm and the defect.
 The absence of the stomach bubble and small bowel in the abdomen supports the diagnosis of CDH.
The absence of the stomach bubble and small bowel in the abdomen supports the diagnosis of CDH.
 Always assess the positions of the umbilical arterial and venous catheters:
Always assess the positions of the umbilical arterial and venous catheters:
Differential Diagnosis
 Multiple cystic structures can be seen within the lung, with contralateral mediastinal displacement.
Multiple cystic structures can be seen within the lung, with contralateral mediastinal displacement.
 There is a more stable position of the abnormality on serial films, whereas this may vary in CDH.
There is a more stable position of the abnormality on serial films, whereas this may vary in CDH.
 Air–fluid levels are more likely in CCAM.
Air–fluid levels are more likely in CCAM.
 If there is greater suspicion of CCAM, a CT examination may be performed, which will show the diaphragm to be intact and confirm the cystic abnormality in the lung.
If there is greater suspicion of CCAM, a CT examination may be performed, which will show the diaphragm to be intact and confirm the cystic abnormality in the lung.
Notes
 Hernias through the foramen of Bochdalek occur posterolaterally with a frequency of between 1 in 2,000 and 1 in 5,000 live births, and are more common on the left side (70%).
Hernias through the foramen of Bochdalek occur posterolaterally with a frequency of between 1 in 2,000 and 1 in 5,000 live births, and are more common on the left side (70%).
 Anterior retrosternal hernias through the foramen of Morgagni are right-sided in 90% of cases.
Anterior retrosternal hernias through the foramen of Morgagni are right-sided in 90% of cases.
 Most neonates present at birth with severe respiratory distress. The sensitivity of ultrasound for the prenatal detection of CDH ranges from 18 to 87%, and appears to increase in the presence of associated, more easily observed anomalies and advancing gestational age.
Most neonates present at birth with severe respiratory distress. The sensitivity of ultrasound for the prenatal detection of CDH ranges from 18 to 87%, and appears to increase in the presence of associated, more easily observed anomalies and advancing gestational age.
 If detected antenatally, fetal endoscopic tracheal occlusion can be performed in selected cases. The procedure is based on the concept that lung growth can be triggered by tracheal occlusion. On the postnatal plain film, a radiodense marker is seen overlying the trachea if the residual balloon has not been removed.
If detected antenatally, fetal endoscopic tracheal occlusion can be performed in selected cases. The procedure is based on the concept that lung growth can be triggered by tracheal occlusion. On the postnatal plain film, a radiodense marker is seen overlying the trachea if the residual balloon has not been removed.
 Abnormalities associated with CDH:
Abnormalities associated with CDH:
Bibliography
6.4 Congenital Lobar Emphysema
Clinical History
Ideal Summary
Examination Tips
 Distribution. Does it involve more than one lobe—the left upper lobe is most commonly affected in CLE.
Distribution. Does it involve more than one lobe—the left upper lobe is most commonly affected in CLE.
 A mass effect can produce a variety of patterns. It is important to be aware of these and adjust your differential accordingly:
A mass effect can produce a variety of patterns. It is important to be aware of these and adjust your differential accordingly:
 Ancillary features:
Ancillary features:
 Attenuated pulmonary vessels in CLE
Attenuated pulmonary vessels in CLE
 Hilar/tubular mass: this is suggestive of bronchial atresia with distended or plugged airways distal to the atretic segment
Hilar/tubular mass: this is suggestive of bronchial atresia with distended or plugged airways distal to the atretic segment
 Small or absent bronchus and pulmonary artery in pulmonary hypoplasia.
Small or absent bronchus and pulmonary artery in pulmonary hypoplasia.
Differential Diagnosis
 Congenital hypoplasia of the lung:
Congenital hypoplasia of the lung:
 Reversible atelectasis:
Reversible atelectasis:
 Bronchial atresia:
Bronchial atresia:
Notes
 Congenital lobar emphysema should be considered when an opacified lobe becomes progressively hyperlucent and hyperinflated just after birth on serial radiographs.
Congenital lobar emphysema should be considered when an opacified lobe becomes progressively hyperlucent and hyperinflated just after birth on serial radiographs.
 The left upper lobe is the most commonly affected, followed by right middle lobe, right upper lobe, and lower lobes. Occasionally, more than one lobe is involved.
The left upper lobe is the most commonly affected, followed by right middle lobe, right upper lobe, and lower lobes. Occasionally, more than one lobe is involved.
 Surgical resection of the affected lobe is indicated, especially in patients with progressive hyperinflation of CLE, which can be life-threatening.
Surgical resection of the affected lobe is indicated, especially in patients with progressive hyperinflation of CLE, which can be life-threatening.
Bibliography
6.5 Hepatoblastoma
Clinical History
Ideal Summary
Examination Tips
 Listen to the examiner for the patient’s age. Hepatoblastoma peaks at 1 to 2 years, with hepatocellular carcinoma (HCC) rare below the age of 3.
Listen to the examiner for the patient’s age. Hepatoblastoma peaks at 1 to 2 years, with hepatocellular carcinoma (HCC) rare below the age of 3.
 Hepatoblastoma is typically large at presentation and is usually single.
Hepatoblastoma is typically large at presentation and is usually single.
 Calcification is not a differentiating feature, being present in 50%.
Calcification is not a differentiating feature, being present in 50%.
 Look for mass effect: hepatoblastoma is more likely to displace rather than invade the hepatic structures.
Look for mass effect: hepatoblastoma is more likely to displace rather than invade the hepatic structures.
 Serum alphafetoprotein is positive in hepatoblastoma (70 to 90% of cases) and HCC (80% of cases), and is negative in haemangioendothelioma.
Serum alphafetoprotein is positive in hepatoblastoma (70 to 90% of cases) and HCC (80% of cases), and is negative in haemangioendothelioma.
Differential Diagnosis
 HCC: This is the most common liver tumour in children over 5 years.
HCC: This is the most common liver tumour in children over 5 years.
 Metastatic disease: This is most commonly from other intra-abdominal primary tumours such as Wilms’ tumour, neuroblastoma, or rhabdomyosarcoma.
Metastatic disease: This is most commonly from other intra-abdominal primary tumours such as Wilms’ tumour, neuroblastoma, or rhabdomyosarcoma.
Notes
 Hepatoblastoma is the most common primary malignant hepatic tumour and accounts for 79% of all liver tumours in children.
Hepatoblastoma is the most common primary malignant hepatic tumour and accounts for 79% of all liver tumours in children.
 Typically, the tumour enhances heterogeneously with intravenous contrast medium, but enhancement is less than the normal liver.
Typically, the tumour enhances heterogeneously with intravenous contrast medium, but enhancement is less than the normal liver.
 High survival rates are seen in those who have complete surgical clearance of the tumour at diagnosis, followed by adjuvant chemotherapy.
High survival rates are seen in those who have complete surgical clearance of the tumour at diagnosis, followed by adjuvant chemotherapy.
 If performing an ultrasound examination:
If performing an ultrasound examination:
 Abnormalities associated with hepatoblastoma:
Abnormalities associated with hepatoblastoma:
Bibliography
6.6 Hirschsprung’s Disease
Clinical History
Ideal Summary
Examination Tips
 Ask for all available views. Early filling anteroposterior and lateral views are essential.
Ask for all available views. Early filling anteroposterior and lateral views are essential.
 The rectum is normally bigger than the sigmoid colon. A rectum: sigmoid ratio of less than 1 is suggestive of Hirschsprung’s disease.
The rectum is normally bigger than the sigmoid colon. A rectum: sigmoid ratio of less than 1 is suggestive of Hirschsprung’s disease.
 The rectum is always involved, but it is important to assess the extent of colonic involvement:
The rectum is always involved, but it is important to assess the extent of colonic involvement:
Differential Diagnosis
 Meconium plug syndrome (also known as functional immaturity of the colon):
Meconium plug syndrome (also known as functional immaturity of the colon):

Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


