Paragangliomas of the head and neck are rare vascular skull-base tumors derived from the paraganglionic system with an estimated incidence of 1:30,000 accounting for 3% of all paragangliomas. The most common paraganglioma locations of the head and neck in descending order are the carotid body, jugular, tympanic, and vagal paragangliomas. This article discusses the clinical characterics, normal anatamy, imaging findings and protocols, pathology, staging, and differential diagnosis for paragangliomas of the head and neck.
Key points
- •
The most common paraganglioma locations of the head and neck are the carotid body, jugular, tympanic, and vagal paragangliomas.
- •
Syndromes associated with a high incidence of paragangliomas include multiple endocrine neoplasia type II, von Hippel-Lindau disease, and neurofibromatosis type I.
- •
All patients, including those with a sporadic tumor, should be referred for a genetic consultation.
- •
Jugular and tympanic paragangliomas commonly present with tinnitus and hearing loss, whereas carotid body and vagal paragangliomas usually present with nonpainful palpable neck mass.
Introduction
Paragangliomas make up a family of neoplasms that develop from the paraganglia tissues, which are themselves of neural crest origin and have similar functions and histologic appearances. They may occur along the ganglia’s pathway of embryologic migration extending from the skull base to the pelvic floor. Paraganglia play an important role in organismic hemostasis by acting directly as chemoreceptors or by the secretion of catecholamines in response to stess. Paragangliomas of the head and neck are rare vascular skull-base tumors derived from the paraganglionic system with an estimated incidence of 1:30,000, accounting for 3% of all paragangliomas.
The most common paraganglioma locations of the head and neck in descending order are the carotid body, jugular, tympanic, and vagal paragangliomas. Tympanic paragangliomas are the most common primary neoplasms of the middle ear. Jugular paragangliomas are the most common tumor of the jugular foramen. Of the different paraganglioma locations, jugulartympanic are the most common paraganglioma causing tinnitus with incidence of 1 per 1.3 million people per year.
Paragangliomas can be sporadic or familial, with genetic mutations occurring in the SDHB, SDHC, or SDHD genes. It is now known that at least 30% of patients with a paraganglioma and no other know risk factors harbor a genetic mutation that increases their risk for these tumors and for other neoplasia. Moreover, other genes discovered within the past 5 years: SDHAF2, TMEM127, MAX, EGLN1, HIF2A, KIF1B, and SDHA, add to the genetic complexity of hereditary paraganglioma-pheochromocytoma syndrome. Sporadic paragangliomas are more common in women, with the average age at presentation of 40 to 50 years, as familial present earlier and more commonly in men. Classic tumor syndromes associated with a high incidence of paragangliomas included multiple endocrine neoplasia type II (MEN II), von Hippel-Lindau (VHL) disease, and neurofibromatosis type I (NF I). Multicentric paragangliomas occur in 10% to 20% of sporadic cases and in up to 80% of hereditary cases.
Paragangliomas are benign neoplasms in most cases; overall, less than 10% of all paragangliomas have been cited to be malignant. In the head and neck, malignancy seems to be more common in vagal paragangliomas (16% to 19%) when compared with carotid body tumors (6%) and jugulartympanic tumors (2% to 4%). There are no accepted histopathological criteria for malignancy, although the diagnosis of a malignant paraganglioma can only be made when there is metastasis to nonneuroendocrine tissue. In head and neck paragangliomas, metastases are most frequently found in cervical lymph nodes; distant sites, which include lung, liver, bone and skin, are very rare.
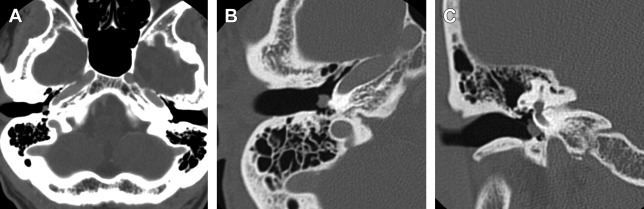
The clinical presentation of a paraganglioma varies based on the tumor location. Jugulartympanic paragangliomas ( Fig. 1 ) present with pulsatile tinnitus (84%) and hearing loss (76%) and frequently cause dysfunction of the cranial nerves VII–XII with large tumors. Tympanic ( Fig. 2 ) and jugular paragangliomas often present to the otolaryngologist as a bluish, pulsating mass behind the eardrum.
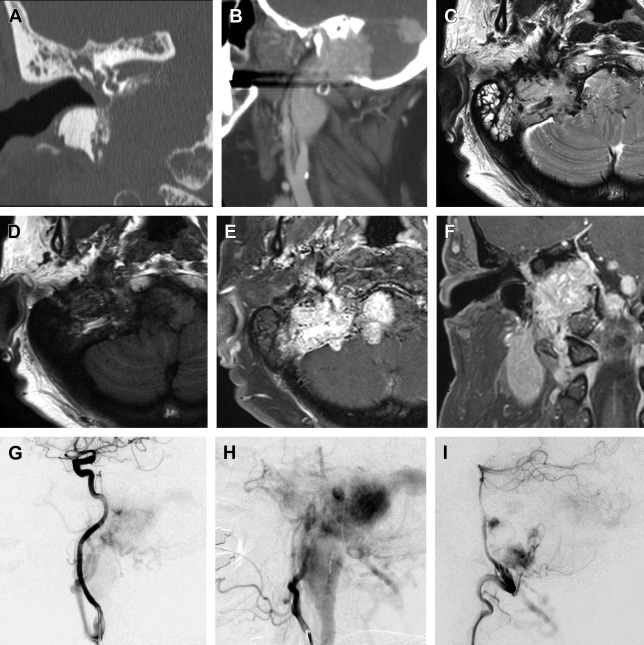
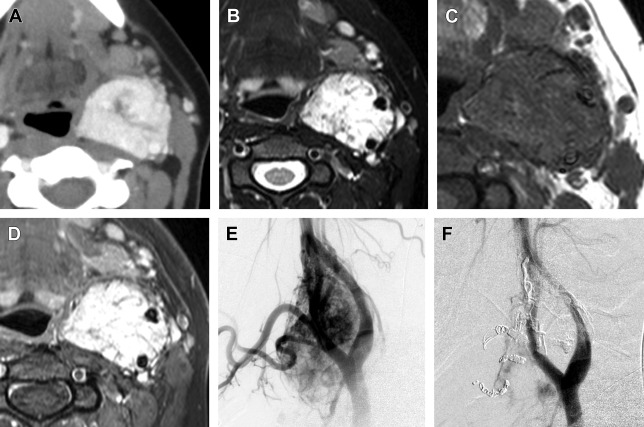
In contrast, carotid body paragangliomas ( Fig. 3 ) present as a painless, slowly enlarging mass lateral to the tip of the hyoid bone. On physical examination, the tumor typically reveals a rubbery, nontender mass along the anterior border of the sternocleidomastoid. The tumor is mobile from side to side, but limited vertically because of the tumor’s attachment to the carotid artery (Fontaine sign). A carotid bruit or pulsatile character of the tumor strengthens the diagnosis. Neurologic deficits of cranial nerves VII, IX, X, XI, and XII can be found in some cases. A painless neck mass is also the most frequent symptom in patients with a vagal paraganglioma ( Fig. 4 ). The tumor most commonly arises from the inferior (nodose) ganglion; however, it can arise from any point along the course of the nerve. Miller and colleagues described their experience with a group of 19 vagal paraganglioma patients. The most frequent presenting symptoms were the presence of a neck mass (n = 14) and hoarseness (n = 7), followed by pharyngeal fullness (n = 5), dysphagia (n = 4), dysphonia (n = 4), pain (n = 4), cough (n = 3), and aspiration (n = 1).
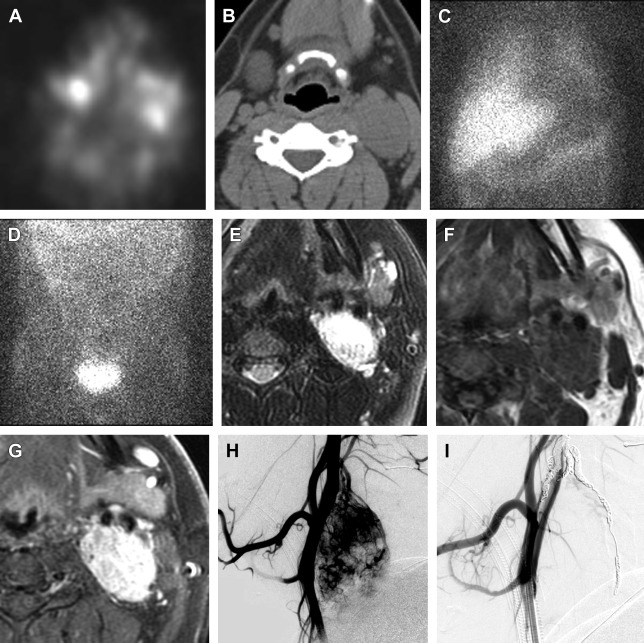
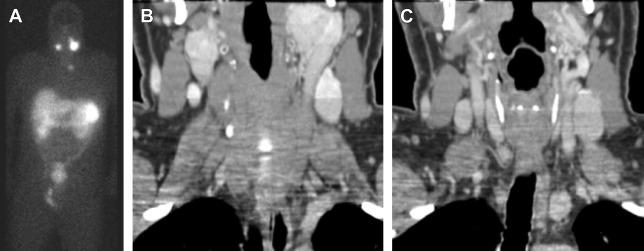
In order to establish a diagnosis of a head and neck paraganglioma, imaging is essential. Depending on the location and extent of the tumor, the following imaging modalities are frequently used: B-mode ultrasonography in combination with color-coded Doppler ultrasonography, computed tomography (CT), MR imaging, and digital subtraction angiography. The most common radioligand for imaging of paragangliomas is In 111-DTPA- d -Phe1-octreotide ( Fig. 5 ).
The different treatment options for paragangliomas include surgical excision, endovascular embolization, conventional radiotherapy, and stereotactic radiosurgery. In selected cases, a watchful approach with repeat imaging may be warranted.
This article discusses normal anatomy and imaging techniques, imaging protocols, imaging findings/pathology, diagnostic criteria, differential diagnosis, and pearls/pitfalls/variants as they relate to paragangliomas involving the skull base. Finally, a brief bulleted list is presented describing what the referring physician needs to know.
Introduction
Paragangliomas make up a family of neoplasms that develop from the paraganglia tissues, which are themselves of neural crest origin and have similar functions and histologic appearances. They may occur along the ganglia’s pathway of embryologic migration extending from the skull base to the pelvic floor. Paraganglia play an important role in organismic hemostasis by acting directly as chemoreceptors or by the secretion of catecholamines in response to stess. Paragangliomas of the head and neck are rare vascular skull-base tumors derived from the paraganglionic system with an estimated incidence of 1:30,000, accounting for 3% of all paragangliomas.
The most common paraganglioma locations of the head and neck in descending order are the carotid body, jugular, tympanic, and vagal paragangliomas. Tympanic paragangliomas are the most common primary neoplasms of the middle ear. Jugular paragangliomas are the most common tumor of the jugular foramen. Of the different paraganglioma locations, jugulartympanic are the most common paraganglioma causing tinnitus with incidence of 1 per 1.3 million people per year.
Paragangliomas can be sporadic or familial, with genetic mutations occurring in the SDHB, SDHC, or SDHD genes. It is now known that at least 30% of patients with a paraganglioma and no other know risk factors harbor a genetic mutation that increases their risk for these tumors and for other neoplasia. Moreover, other genes discovered within the past 5 years: SDHAF2, TMEM127, MAX, EGLN1, HIF2A, KIF1B, and SDHA, add to the genetic complexity of hereditary paraganglioma-pheochromocytoma syndrome. Sporadic paragangliomas are more common in women, with the average age at presentation of 40 to 50 years, as familial present earlier and more commonly in men. Classic tumor syndromes associated with a high incidence of paragangliomas included multiple endocrine neoplasia type II (MEN II), von Hippel-Lindau (VHL) disease, and neurofibromatosis type I (NF I). Multicentric paragangliomas occur in 10% to 20% of sporadic cases and in up to 80% of hereditary cases.
Paragangliomas are benign neoplasms in most cases; overall, less than 10% of all paragangliomas have been cited to be malignant. In the head and neck, malignancy seems to be more common in vagal paragangliomas (16% to 19%) when compared with carotid body tumors (6%) and jugulartympanic tumors (2% to 4%). There are no accepted histopathological criteria for malignancy, although the diagnosis of a malignant paraganglioma can only be made when there is metastasis to nonneuroendocrine tissue. In head and neck paragangliomas, metastases are most frequently found in cervical lymph nodes; distant sites, which include lung, liver, bone and skin, are very rare.
The clinical presentation of a paraganglioma varies based on the tumor location. Jugulartympanic paragangliomas ( Fig. 1 ) present with pulsatile tinnitus (84%) and hearing loss (76%) and frequently cause dysfunction of the cranial nerves VII–XII with large tumors. Tympanic ( Fig. 2 ) and jugular paragangliomas often present to the otolaryngologist as a bluish, pulsating mass behind the eardrum.
In contrast, carotid body paragangliomas ( Fig. 3 ) present as a painless, slowly enlarging mass lateral to the tip of the hyoid bone. On physical examination, the tumor typically reveals a rubbery, nontender mass along the anterior border of the sternocleidomastoid. The tumor is mobile from side to side, but limited vertically because of the tumor’s attachment to the carotid artery (Fontaine sign). A carotid bruit or pulsatile character of the tumor strengthens the diagnosis. Neurologic deficits of cranial nerves VII, IX, X, XI, and XII can be found in some cases. A painless neck mass is also the most frequent symptom in patients with a vagal paraganglioma ( Fig. 4 ). The tumor most commonly arises from the inferior (nodose) ganglion; however, it can arise from any point along the course of the nerve. Miller and colleagues described their experience with a group of 19 vagal paraganglioma patients. The most frequent presenting symptoms were the presence of a neck mass (n = 14) and hoarseness (n = 7), followed by pharyngeal fullness (n = 5), dysphagia (n = 4), dysphonia (n = 4), pain (n = 4), cough (n = 3), and aspiration (n = 1).
In order to establish a diagnosis of a head and neck paraganglioma, imaging is essential. Depending on the location and extent of the tumor, the following imaging modalities are frequently used: B-mode ultrasonography in combination with color-coded Doppler ultrasonography, computed tomography (CT), MR imaging, and digital subtraction angiography. The most common radioligand for imaging of paragangliomas is In 111-DTPA- d -Phe1-octreotide ( Fig. 5 ).
The different treatment options for paragangliomas include surgical excision, endovascular embolization, conventional radiotherapy, and stereotactic radiosurgery. In selected cases, a watchful approach with repeat imaging may be warranted.
This article discusses normal anatomy and imaging techniques, imaging protocols, imaging findings/pathology, diagnostic criteria, differential diagnosis, and pearls/pitfalls/variants as they relate to paragangliomas involving the skull base. Finally, a brief bulleted list is presented describing what the referring physician needs to know.
Normal anatomy and imaging technique
Normal Anatomy
The 3 most common paragangliomas of the head and neck are the carotid body, jugular, and tympanic tumors. These tumors are located in the following areas: the carotid body paraganglioma is located at the carotid bifurcation; the jugular paraganglioma is located at the jugular foramen/bulb; and the tympanic paraganglioma is located at the cochlear promontory. Therefore, paragangliomas of the head and neck require extensive anatomic knowledge of the jugular foramen, middle ear, and anterolateral neck.
Jugular fossa (JF) anatomy is important for understanding glomus jugular and tympanic paraganglioma pathology and imaging. The JF is a deep depression located in the temporal bone and cradles the jugular bulb. The JF is located directly behind the entrance of the carotid canal, lateral to the anterior half of the occipital condyle, medial to the stylomastoid foramen, posteromedial to the styloid process, and posteroinferior to the middle ear cleft. The foramen can be divided into anteromedial as well as posterolateral compartments. The anteromedial compartment contains the glossopharyngeal nerve, inferior petrosal sinus, and meningeal branch of the ascending pharyngeal artery. The posterolateral compartment contains the vagus and spinal accessory nerves. Tympanic paragangliomas typically arise from the tympanic plexus formed by tympanic nerve (Jacobson nerve), a branch of the glossopharyngeal nerve. The plexus of nerves lies on the cochlear promontory of the middle ear. The inferior tympanic artery, which is a branch of the ascending pharyngeal artery, accompanies the nerve and usually also occupies a groove in the promontory.
Carotid body paragangliomas originate from the glomus body at the carotid bifurcation between the external carotid artery (ECA) and the internal carotid artery (ICA). Thus, it is important to understand the neurovascular relationship of the ICA as well as the ECA branches in the anterolateral neck. Nerves that pass between the ICA and ECA are at risk during surgery, including the glossopharyngeal nerve, superior laryngeal nerve (vagal branch), and pharyngeal nerve (vagal branch). However, with extension of the tumor, the hypoglossal nerve and the mandibular nerve (facial branch) may also be involved.
Imaging Technique
Along with medical history and targeted physical examination, imaging plays an important role in the diagnosis and management of head and neck paragangliomas. Initial imaging is usually done with CT and/or MR imaging. Angiography is usually performed after the diagnosis is established for preoperative assessment. Functional imaging with nuclear medicine radiotracers is used to evaluate for multifocal disease or distant metastases, but is not standard practice.
The initial imaging modality of choice to assess temporal bone involvement and to visualize bony structures and tumor extension is a high-resolution CT (HRCT) scan (<1 mm) in the axial, direct tumor plane, and reconstructed coronal plane. However, when a suspicious mass is identified on a noncontrast HRCT, a contrast-enhanced temporal bone CT can help differentiate an intensely enhancing paraganglioma from other benign and malignant tumors of the skull base. Compared with HRCT, multiplanar MR imaging with T1- and T2-weighted images along with gadolinium-enhanced images provides superior soft tissue details and identification of intracranial tumor extension.
The imaging modality of choice for preoperative planning and tumor characterization is angiography. Angiography can identify the highly vascular tumor and provide important preoperative vascular mapping before surgical excision, and when indicated, embolization of the dominant arterial feeders can be performed before tumor resection. Furthermore, angiography can characterize collateral vessels that must be spared during surgery and can provide further information about the patency of the venous system and multifocal nature of the tumor.
Staging of the tumor for multifocal or distant metastases is done with nuclear medicine imaging and is becoming more sensitive and specific, but is not standard practice yet. The most common radioligand is In 111-DTPA- d -Phe1-octreotide, which has been shown to be superior to I-131 and I-123 metaiodobenzylguanidine (MIBG) in clinical trials. However, novel radiotracer F-18 FDOPA (fluorodopa) PET/CT is highly sensitive (>95%) and specific (95%–100%) for head and neck paraganglioma detection, but is not readily available at most sites. Rapidly emerging radiotracer Ga-68 SSTa (Ga-68-labeled somatostatin analogues) combined with PET/CT could evolve as the new imaging standard of reference for imaging of paragangliomas and does not require a cyclotron to make the tracer.
Imaging Protocols
CT and MR imaging protocols for imaging jugular, tympanic, and carotid body paragangliomas are included in Tables 1–6 .
| Examination Code MR70553 | Series Description | Sag T1 TSE | AX DWI | AX T1 FLAIR | AX T2 STAR | AX T2 TSE |
|---|---|---|---|---|---|---|
| Patient | Patient position | Supine | Supine | Supine | Supine | Supine |
| Position | Patient orientation | Head first | Head first | Head first | Head first | Head first |
| Coil type | Base/HN | Base/HN | Base/HN | Base/head | Base/HN | |
| Slice orientation | Sagittal | Axial | Axial | Axial | Axial | |
| Coverage | — | Whole brain | Whole brain & CN V | Whole brain | Whole brain | Top of frontal sinus to C3 including mandible |
| Geometry | Field of view | 230 | 256 | 254 × 200 | 254 × 200 | 160 |
| Voxel size | FH 0.78 | RL 1.5 | AP 0.69 | AP 0.9 | AP 0.60 | |
| AP 0.78 | AP 1.5 | RL 0.94 | RL 1.12 | RL 0.60 | ||
| Slice thickness | 5/1 | 5/1 | 5/1 | 5/1 | 4/1 | |
| Sense factor | 1.6 | 4 | 1.5 | 1.8 | 2 | |
| No. slices | 21 | 25 | 25 | 25 | 34 | |
| Fold-over direction | AP | AP | RL | RL | RL | |
| Contrast | Scan mode | MS | MS | MS | MS | MS |
| Technique | IR | SE | IR | FFE | SE | |
| Fast imaging mode | TSE | EPI | TSE | — | TSE | |
| TSE factor | 5 | SS | 27 | — | 15 | |
| TE | 10 | Shortest | 125 | In-phase 16.11 | 80 | |
| TR | Shortest | Shortest | 11,000 | Shortest | Shortest | |
| Flip angle | — | 90 | — | 18 | 90 | |
| Fat saturation | — | SPIR | SPIR | — | — | |
| Motion | NSA | 1 | 2 | 1 | 1 | 2 |
| Scan time | Scan duration | 3:42 | 2:45 | 2:45 | 1:19 | 2:21 |
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


