Fig. 9.1
Structure and function of the aortic valve. (a) Pressure generated in the left ventricle during systolic contraction forces the aortic valve leaflets into the aorta, allowing blood to be ejected from the heart. (b) As the heart rests during diastole, the leaflets strain radially to meet and close the valve, preventing retrograde blood flow into the ventricles. The circumferentially aligned collagen fibers give the leaflets strength to prevent prolapse. (c) The aligned collagen fibers are shown using polarized light imaging of picrosirius red staining. Original magnification ×200 (Figure adapted from Hutcheson et al. [14])
The leaflets must simultaneously exhibit the compliance necessary for opening during systole and the biomechanical strength to resist prolapse back into the left ventricle during diastole [2]. This is accomplished through a layered leaflet microarchitecture that is crucial to ensure proper valve function [3]. The aortic side of the leaflets, a region known as the fibrosa layer, is composed of circumferentially aligned collagen fibers that straighten during diastole and serve as the main tensile load bearing component of the leaflets (Fig. 9.1c) [4]. On the ventricular side of the leaflets, an elastin layer, known as the ventricularis, confers radial elasticity that allows the valve to open during systole and allows the leaflets to stretch radially and meet to close the valve during diastole [5]. An intermediary layer, known as the spongiosa, is enriched in glycosaminoglycans and is believed to serve as a lubricating layer between the fibrosa and ventricularis. This microarchitecture is maintained by two resident cell populations: the aortic valve endothelial cells (VECs) that sheath the leaflets and the aortic valve interstitial cells (VICs) that reside within the leaflets. Inflammation and pathological differentiation of these resident cells have been shown to lead to calcific aortic valve disease (CAVD). Histopathological analyses of CAVD reveal fibrotic collagen accumulation and the formation of calcific nodules on the aortic side of the leaflets [6]. These changes disrupt the microarchitecture of the leaflets and thereby compromise the biomechanical integrity of the aortic valve. The following sections detail the cellular and molecular changes observed in CAVD and the role that molecular imaging may play in both research and clinical intervention.
9.2 Imaging Criteria for Aortic Valve Disease
CAVD is a major contributor of cardiovascular morbidity and mortality. Of particular concern, the prevalence of CAVD is projected to increase dramatically due to the shift in age-related population demographics with an aging baby boomer generation. Moderate-to-severe symptoms of CAVD are observed in 2.8 % of patients over 75 years of age [7, 8]. Given this strong association with age, for many years, CAVD was presumed to be a passive, degenerative disease that occurs due to fatigue or a wearing out of the tissue caused by the stresses and strains of cardiac function as the aortic valve opens and closes more than three billion times over the course of an average life span [9]. Recently, however, the results of many studies on the molecular signaling processes associated with valvular homeostasis have led to a paradigm shift with CAVD now viewed as an active, cellular-driven disease [10]. This view offers some hope to patients as cellular processes may be targeted therapeutically; however, no clinically relevant therapeutic options for CAVD have been discovered.
The development of a therapeutic for CAVD dovetails with the need for detection strategies to identify early changes within the leaflets [11]. Once calcium deposits form within the valve, reversibility is unclear; however, inflammation and the early extracellular matrix remodeling responses that presage gross fibrosis and calcific nodule formation may fall within an appropriate therapeutic window (Fig. 9.2). Traditional detection of valvular insufficiency using echocardiography is unable to identify these early leaflet changes as regurgitant blood flow and leaflet thickening represent later stages of CAVD remodeling and calcification. Recently, PET/CT imaging modalities have emerged as a means to detect inflammation and mineralization within valve leaflets of human patients [12], and optical molecular imaging strategies have been successfully employed to visualize the progression of CAVD in mice [13]. These technologies are far more sensitive to small changes within the leaflets and may offer the resolution necessary to identify patients that fall within the appropriate therapeutic time frame. Utilizing these imaging strategies may also give better insight into the temporal aspects of CAVD and may lead to a better understanding of the required therapeutic strategy at each stage of CAVD.
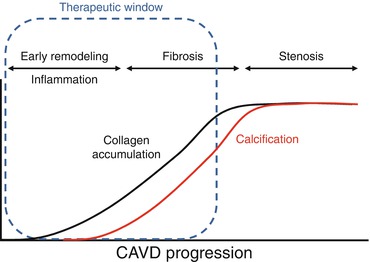
Fig. 9.2
Early tissue remodeling and inflammation is thought to precede gross fibrosis and calcification within CAVD leaflets. Calcification may be an irreversible endpoint; therefore, identifying patients in the early stages of CAVD may present the best opportunity for therapeutic intervention
The reader should note that many of the processes and techniques discussed in this chapter overlap with those discussed in Chap. 5 . Many of the pathophysiological changes in CAVD and vascular calcification overlap; however, it should be noted that these diseases are now believed to have distinct etiologies [14]. Not every patient who presents prominent vascular calcification also exhibits signs of CAVD, suggesting that at least an extra set of variables are involved in CAVD initiation and progression. Further, valve cells are phenotypically different than their vascular counterparts, and therefore, different factors seem to be responsible for producing the fibrocalcific endpoint that is apparent in both diseases. The goal of molecular imaging for these two diseases is also different. In vascular calcification, a collagen-rich fibrous cap with a large calcification may serve to stabilize the atherosclerotic plaque [15, 16]; therefore, the first goal in imaging of atherosclerotic lesions is to identify regions of collagen disruption and the formation of spotty microcalcifications that lead to plaque instability. Conversely, in CAVD, aortic valve leaflet microarchitecture is essential to the appropriate biomechanical function of the valve. Therefore, molecular imaging tools should be designed around early identification of valve remodeling prior to the potentially irreversible endpoints of fibrosis and calcification. In this chapter, we will focus on the preclinical studies that have exhibited promise in imaging processes involved in CAVD and the novel findings that have resulted from these imaging modalities. We will also use our current understanding of the molecular underpinnings of CAVD to propose potential new targets for imaging that might distinguish CAVD from vascular calcification. Finally, we will discuss how molecular imaging techniques may be translated to clinical use to identify early markers of CAVD in patients.
9.3 Valve Endothelial Cell Activation and Migration
In other parts of the vascular system, endothelial cells have been observed to play a crucial role in the transduction of mechanical signals such as changes in shear stress [17]. Because these cells are the first to experience any changes in outside forces that affect valve dynamics, they have received growing attention in their potential roles in regulating valve homeostasis. VECs are phenotypically distinct from endothelial cells in the vasculature [18]. For instance, when subjected to the shear stresses of fluid flow, VECs align perpendicular to the flow [18]; whereas, endothelial cells from the aorta align parallel to the flow [17, 19]. This indicates that the VECs may play a distinct role in regulating the biomechanical properties of the valve through specially evolved mechanisms. Furthermore, VECs have been observed to exhibit genotypic heterogeneity along the surface of each leaflet. Simmons et al. found that 584 genes were differentially expressed between VECs from the aortic side and the ventricular side [20]. These genotypic differences may be important in regulating the differing biomechanical properties on each side of the valve, i.e., the collagen of the fibrosa and the elastin of the ventricularis, or conversely, the gene expression of these cells may be influenced by the differing shear stresses seen on each side of the leaflet.
VECs have also been shown to play a role in the etiology and progression of valve disease. In diseased or injured vascular tissues, endothelial cells direct the appropriate immune response through increased expression of cell adhesion molecules (CAMs) that mediate attachment and activation of leukocytes as well as by secreting cytokines that are recognized by specialized inflammatory cells [21–24]. Similarly, VECs have been observed to increase expression of ICAM-1 and VCAM-1 and secretion of inflammatory cytokines such as IL-8, IL-1β, and IFN-γ in response to oscillatory, turbulent fluid flows and pathological mechanical strains [25]. As discussed in the following section, the accumulation of leukocytes and related inflammatory cytokines is positively correlated with remodeling in CAVD leaflets and aortic stenosis in human patients. Ex vivo MRI and near-infrared fluorescence (NIRF) imaging of a VCAM-1-targeted agent demonstrated VEC activation localized in regions where the valve leaflets meet the aortic root [26] (Fig. 9.3). Correspondingly, this area of the leaflet—known as the commissure—experiences the greatest amount of mechanical stress and, therefore, is most likely to experience VEC damage that leads to CAM expression [27].
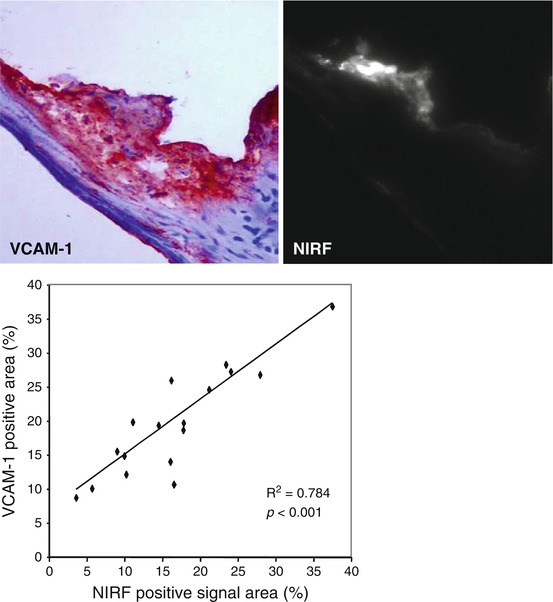
Fig. 9.3
A NIRF probe targeted to VCAM-1 was detected in the commissure region of apoE-deficient mouse aortic valves. The specificity of this probe was validated and quantified using traditional immunohistological staining and direct visualization of VCAM-1 activity using NIRF microscopy (Figure modified Aikawa et al. [13])
VECs may also play an important, though lesser known, function in CAVD, and the spatial heterogeneity of these cells could be responsible for the side-specific susceptibility to CAVD observed in histopathological analyses of diseased leaflets [20]. The VECs directly adjacent to the fibrosa have been found to express much lower levels of anti-calcification enzymes when compared to the VECs on the ventricular side of the leaflets [25]. This suggests that the VECs may also play an important role in sensing and responding to the various stresses on each side of the leaflets. Recent studies have suggested that VECs play an active role in valve remodeling during CAVD by invading the leaflet interstitium through endothelial-to-mesenchymal (EMT) processes that mirror those from development [28, 29]. The addition of mechanical strain to either valve leaflets in vivo or an in vitro culture system of VECs led to the expression of mesenchymal markers such as smooth muscle α-actin (α-SMA) within the VECs [28] (Fig. 9.4). VECs isolated from ovine mitral valve leaflets were shown to exhibit a propensity for osteogenic and chondrogenic differentiation that was not found in vascular endothelial cells [30]. Markers of differentiation were exacerbated by exposing leaflets to mechanical strain. Therefore, future high-resolution imaging techniques may be able to search for localization of VEC marker proteins within the interstitial space or mesenchymal markers on the leaflet surface as early signs of CAVD.
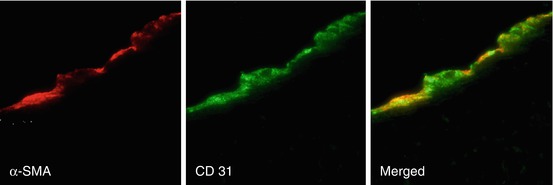
Fig. 9.4
The addition of mechanical strain to excised mitral valve leaflets showed an increase in mesenchymal markers such as α-SMA in the endothelium (identified by CD31 staining) (Images adapted from Wylie-Sears et al. [30])
9.4 Macrophages and Proteolytic Activity
Activation of VECs leads to the recruitment of monocytes and other leukocytes to the valve leaflets. As a result, CAVD (similar to calcification in atherosclerotic plaques) is hallmarked by the presence of inflammatory cells, such as macrophages and T cells, within and surrounding calcified regions of the tissue [12, 31–33]. Molecular imaging techniques using macrophage-targeted iron nanoparticles conjugated to a NIRF agent identified a strong correlation between macrophage accumulation and calcification in aortic valves of apolipoprotein E-deficient (apoE−/−) mice [34] (Fig. 9.5). These cells may play a role in initiating the remodeling processes that lead to calcification, as they actively release proteases that degrade extracellular matrix components, priming the tissue for new matrix secretion. Additionally, the accumulation of leukocytes and related inflammatory cytokines is positively correlated with remodeling in CAVD leaflets and aortic stenosis in human patients. Similarly, in an apoE−/− mouse model of atherosclerosis and CAVD, enzymatically activatable NIRF signal corresponding to a specific protease cathepsin B (as validated by immunohistological techniques) was shown to co-localize with macrophages within the valve leaflets (Fig. 9.6) [13].
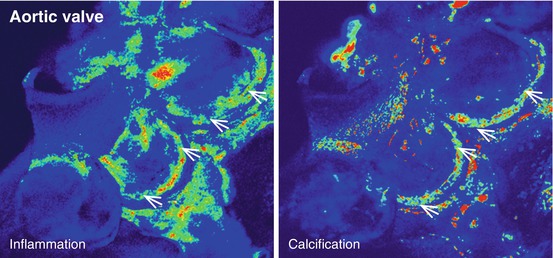
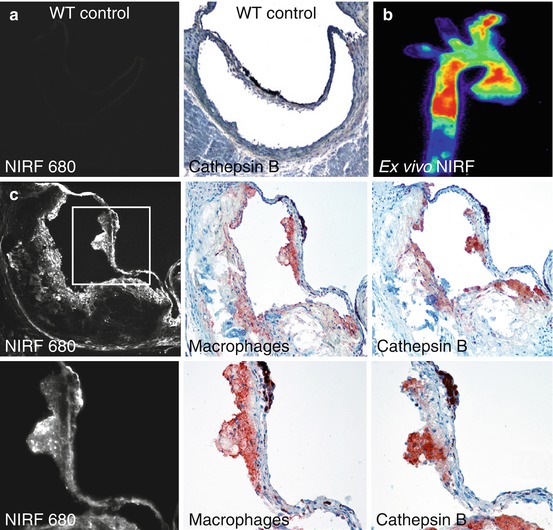
Fig. 9.5
Ex vivo NIRF imaging of the aortic valve of an apoE−/− mouse revealed a strong association between the presence of inflammation and calcification within the leaflets (Adapted from Aikawa et al. [34])
Fig. 9.6
Macrophage activity in the aortic valve. (a) Wild-type (WT) control mice did not exhibit detectable cathepsin B activity within the aortic valve. (b) ApoE−/− mice showed strong cardiovascular NIRF signal from a cathepsin B activatable probe. (c) Immunohistological analyses of the aortic valve showed a co-localization between the cathepsin NIRF signal and cathepsin B expression in macrophages within the valve (Modified from Aikawa et al. [13])
Other members of the cathepsin family are involved in elastin degradation. As mentioned in the first section, the ventricularis layer of the aortic valve is composed mainly of elastin that allows the valve leaflets to exhibit a high radial strain response to diastolic loading. Disruption of this elastin layer has been shown to result in pathological remodeling associated with CAVD [34–36]. Chronic renal disease (CRD) is strongly correlated with CAVD in human patients, and CAVD processes in apoE−/− mice are accelerated by inducing CRD through resection of 5/6 of the total kidney volume (5/6 nephrectomy). Interestingly, induction of CRD in apoE−/− mice leads to a marked increase in activity of the elastolytic enzyme cathepsin S as shown by an enzymatically activatable NIRF agent. Deletion of cathepsin S in these mice mitigates CAVD progression [34]. Cathepsin S remodeling in this mouse model was found to be closely associated with the presence of inflammatory cell markers within the leaflets. In addition, elastin degradation peptides, also known as matrikines, highly biologically active molecules, may induce VIC osteogenic differentiation in aortic valves [34]. Application of pathological cyclic strain to aortic valve leaflets in an ex vivo bioreactor demonstrated significant increases in activity of cathepsin S and a similar protease, cathepsin K, within the leaflets.
9.5 Myofibroblast Activation, Osteogenic Differentiation, and Calcification
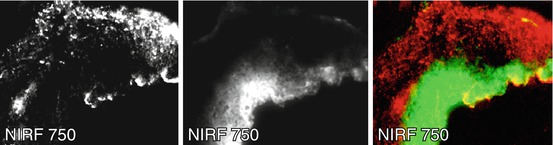
As noted in the vessel wall, molecular imaging of calcific mineral deposition has been achieved using bisphosphonates conjugated to NIRF molecules [37]. Bisphosphonates have a structure similar to the calcification inhibitor pyrophosphate [38] and is believed to bind to calcium and accumulate in mineralized crystals [39]. The accumulation of NIRF-bisphosphonates in calcific regions of CAVD leaflets yields detectable NIRF signal that is spectrally distinct from the protease agents discussed in the previous section, allowing for simultaneous imaging of these processes [40] (Fig. 9.7).
 Molecular Imaging of Macrophages in Atherosclerosis
Molecular Imaging of Macrophages in Atherosclerosis
 Molecular Imaging of Oxidation-Specific Epitopes to Detect High-Risk Atherosclerotic Plaques
Molecular Imaging of Oxidation-Specific Epitopes to Detect High-Risk Atherosclerotic Plaques
 Ultrasound Imaging of Calcific Aortic Valve Disease
Ultrasound Imaging of Calcific Aortic Valve Disease
 Pathobiology and Mechanisms of Atherosclerosis
Pathobiology and Mechanisms of Atherosclerosis
 PET/CT Imaging of Inflammation and Calcification in CAVD: Clinical Studies
PET/CT Imaging of Inflammation and Calcification in CAVD: Clinical Studies
 Molecular MR Imaging of Atherosclerosis
Molecular MR Imaging of Atherosclerosis




Related posts:
Molecular Imaging of Macrophages in Atherosclerosis
Molecular Imaging of Oxidation-Specific Epitopes to Detect High-Risk Atherosclerotic Plaques
Ultrasound Imaging of Calcific Aortic Valve Disease
Pathobiology and Mechanisms of Atherosclerosis
PET/CT Imaging of Inflammation and Calcification in CAVD: Clinical Studies
Molecular MR Imaging of Atherosclerosis
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
