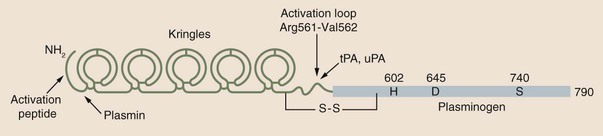
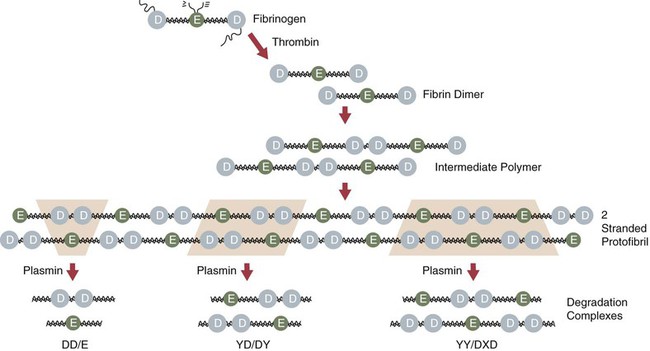
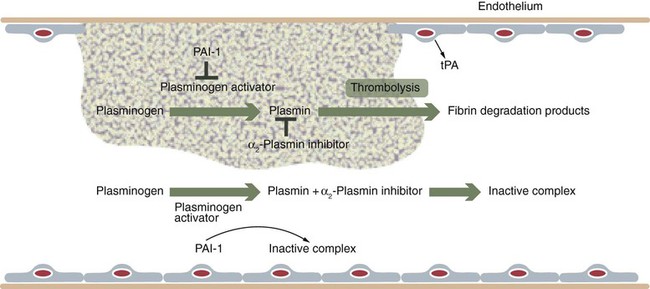
Chapter 21 Michael B. Streiff and Raagsudha Jhavar The fibrinolytic system is a critical regulatory network of proteins that provide balance to the hemostatic mechanism by ensuring maintenance of vascular patency by opposing the procoagulant forces of the coagulation cascade proteins. The key fibrinolytic enzyme in blood is plasmin, a serine protease that circulates as the inactive zymogen, plasminogen. Although plasminogen expression has been identified in a broad range of tissues, including the adrenal glands, brain, heart, intestine, kidney, lung, spleen, testis, thymus, and uterus, plasminogen is primarily synthesized in the liver. Plasminogen is a 92-kD, 791–amino acid glycoprotein whose plasma concentration in normal adults is 200 mg/L.1–3 The concentration of plasminogen in premature infants and newborns, respectively, is 75% and 50% lower than in adults.4 The half-life of plasminogen is 2.2 days, primarily because of catabolic degradation rather than consumption via conversion to plasmin. Although plasmin/plasminogen is primarily known for its function in the dissolution of fibrin thrombi, it also plays a key role in digestion of proteins critical for cellular migration, angiogenesis, and wound healing. Homozygous deficiency of plasminogen is associated with ligneous conjunctivitis, hydrocephalus, and abnormalities in wound healing, defects that can be corrected with plasminogen replacement therapy. Despite its role in thrombolysis, plasminogen deficiency or dysfunction has not been associated with an increased risk for thrombosis in affected patients.1,5 The N-terminal activation peptide is so named because cleavage of it (usually promoted by plasmin) from full-length Glu1-plasminogen results in Lys78-plasminogen that exists in a relaxed conformation that is more easily activated by plasminogen activators. The five kringle domains consist of 80–amino acid loops held together by three disulfide bridges that allow plasminogen to bind to fibrin/fibrinogen, bacterial proteins, cell surfaces, endogenous inhibitors such as α2-antiplasmin, and antifibrinolytic medications such as ε-aminocaproic acid and tranexamic acid. Homologous structures are found in tissue plasminogen activator (tPA), urokinase plasminogen activator (uPA), prothrombin, lipoprotein (a), factor XIII, and hepatocyte growth factor. Binding of fibrinogen converts plasminogen to a more relaxed conformation that facilitates activation of it by plasminogen activators and mediates the concentration of reactants (plasminogen, plasminogen activator, and fibrin) to form a ternary complex that increases the efficiency of fibrinolysis. Adjacent to the kringle domains is the activation or C (cleavage) loop, a 39–amino acid structure that contains the Arg561-Val562 sequence, which is cleaved by plasminogen activators to generate the active enzyme plasmin. The C-terminal end of plasminogen contains the serine protease active site that cleaves target substrate proteins such as fibrin.1,2,5–8 Conversion of plasminogen to plasmin by plasminogen activators is mediated by cleavage of the Arg561-Val562 bond in the C loop of plasminogen (Fig. e21-1). This event creates a two-chain structure composed of a heavy (or A) chain of 561 amino acids that contains the amino-terminal kringle domains and a light (B) chain of 230 amino acids that contains the carboxy-terminal active site. Plasminogen is also digested by plasmin into a truncated form of plasmin known as Lys78-plasminogen, which lacks the N-terminal 77 amino acids that are present in the full-length form of plasminogen known as Glu1-plasminogen (see Fig. e21-1). Both Glu1- and Lys78-plasminogen can be converted by plasminogen activators to plasmin; however, Lys78-plasminogen appears to be activated at a higher rate than Glu1-plasminogen in the absence of fibrin.1,2,6,8 The carboxy-terminal active site of plasmin is formed by a catalytic triad of three amino acid residues, His602, Asp645, and Ser740, and it shares considerable homology with other serine proteases, including trypsin, thrombin, and factor Xa. As a serine protease, it cleaves peptide bonds containing lysine or arginine on the carboxy side of the molecule. The principal physiologic substrate for plasmin degradation is fibrin/fibrinogen, although plasmin has also been demonstrated to degrade other coagulation factors, including factors Va and VIIIa, as well as adhesive proteins such as von Willebrand factor, the platelet membrane receptors glycoprotein IIb/IIIa and glycoprotein lb, and basement membrane proteins such as laminin, fibronectin, and thrombospondin. Cleavage of fibrin by plasmin generates a series of cross-linked fragments, including D-dimers, DD/E complexes, and combinations of D, X, and Y fragments that are measurable in plasma (Fig. e21-2). These fragments can impede fibrin polymerization and, in conjunction with the degradation of fibrinogen factors Va and VIIIa and platelet glycoprotein receptors, contribute to the antithrombotic properties of plasmin.1,2,6–8 α2-Antiplasmin is synthesized by the liver and has a plasma concentration of 7 mg/dL. The plasma half-life of α2-antiplasmin is 2.6 days. Its half-life decreases to 0.5 day when complexed to plasmin. Although α2-antiplasmin is primarily a plasma protein, platelets also contain a small amount of α2-antiplasmin in alpha granules that is released upon platelet activation. α2-Antiplasmin circulates in two forms in the plasma, the native unprocessed form synthesized by the liver and a processed form that undergoes cleavage by antiplasmin-cleaving enzyme at the Pro12-Asm 13 bond. The processed form of α2-antiplasmin binds to fibrin 13-fold more rapidly, providing greater protection against fibrinolysis by plasmin. α2-Antiplasmin rapidly inhibits plasmin in a two-step process, initially by forming a reversible 1 : 1 stoichiometric complex with plasmin. Subsequently, cleavage of the Arg364-Met365 bond in the active site of α2-antiplasmin leads to formation of a covalent bond between Arg364 (of α2-antiplasmin) and Ser740 in the active site of plasmin, thereby generating an irreversible inhibitory complex.2,3,6,7 The physiologic significance of α2-antiplasmin is underscored by the clinical symptoms suffered by patients with homozygous α2-antiplasmin deficiency. This rare bleeding disorder is inherited in an autosomal recessive fashion and is characterized by bleeding comparable in severity to that of hemophilia A. The bleeding typically occurs in delayed fashion after initial hemostasis, consistent with defective regulation of fibrinolysis. Laboratory testing demonstrates severely reduced levels of α2-antiplasmin and a rapid euglobulin lysis time. Patients suffering from heterozygous deficiency of α2-antiplasmin have also been rarely described, and some have suffered from a mild bleeding tendency. Symptomatic patients with α2-antiplasmin can be successfully managed with antifibrinolytic agents such as ε-aminocaproic acid. Acquired causes of α2-antiplasmin deficiency include disseminated intravascular coagulation, thrombolytic therapy, liver disease (secondary to impaired production), nephrotic syndrome (secondary to excretion), and systemic amyloidosis.6 α2-Macroglobulin, a 725-kD glycoprotein synthesized by a variety of cells including hepatocytes and macrophages, plays a secondary role in the regulation of plasmin. α2-Macroglobulin does not play a primary role in plasmin inhibition until α2-antiplasmin is consumed. The plasma concentration of α2-macroglobulin is 2 mg/mL. Unlike α2-antiplasmin, α2-macroglobulin does not inhibit plasmin by binding to its active site but rather by sequestering it. When the target protease cleaves a trigger peptide sequence in α2-macroglobulin, a conformational change occurs that entraps the protease. The α2-macroglobulin-protease complex is then rapidly cleared from the circulation by receptor-mediated endocytosis, primarily in the liver.2,6,7 Binding to vitronectin and fibrin can stabilize PAI-1 in the active form. Transition to the latent form of PAI-1 occurs spontaneously in a time-dependent process. PAI-1 inhibits tPA and uPA in a 1 : 1 molar ratio by forming a covalent bond between its inhibitory reactive center and the active site serine of the target plasminogen activator.2,3,6–8 Synthesis of PAI-1 is induced by a diverse group of substances including insulin, endotoxin, thrombin, and atherogenic lipoproteins and cytokines. Elevated levels of PAI-1 have been noted in association with sepsis, malignancy, pregnancy, the postoperative state, and acute thrombosis. These changes may contribute to the excess risk for thrombosis associated with these clinical states. The 4G/5G polymorphism in the PAI-1 promoter has been demonstrated to influence PAI-1 transcription rates, with individuals possessing the 4G/4G allele having higher PAI-1 levels. Some but not all studies have found an association between higher PAI-1 levels and venous thromboembolism. Conversely, rare individuals with PAI-1 deficiency have manifested a bleeding disorder.2,3,6,7 A second negative regulator of plasminogen activation, PAI-2, has also been identified. PAI-2 is generally found in detectable quantities only during pregnancy and appears to be synthesized by placental villous cells. The increase in plasma concentrations of PAI-2 probably contributes to the hypofibrinolytic state associated with pregnancy.2,6,8 Other slower-acting inhibitors of tPA and uPA include α1-antitrypsin, α2-macroglobulin, α2-antiplasmin, C1-inhibitor, and PAI-3.8 These residues are high-affinity binding sites for plasminogen and tPA that are generated with the initiation of fibrinolysis and explain the acceleration of fibrinolysis that occurs after initial lysis starts. Removal of these residues reduces plasminogen and tPA fibrin binding, and the conversion of Glu-plasminogen to Lys-plasminogen that prevents acceleration of fibrinolysis. TAFI is a 58-kD, 401–amino acid glycoprotein that is synthesized by the liver. TAFI is also released locally at the site of clot formation from platelet alpha granules during platelet activation. The mean plasma concentration of TAFI is 200 nM.7,8 The severely attenuated thrombin burst generated by hemophiliac plasma not only contributes to less fibrin generation but also activates less activated TAFI and factor XIII, such that the fibrin clot is more susceptible to subsequent fibrinolysis. TAFI levels increase with age and in women taking hormonal therapy, although pregnancy has no effect on TAFI levels. Decreased TAFI levels have been noted in patients with severe liver disease and acute promyelocytic leukemia and may contribute to the bleeding diathesis seen in these conditions. Increased levels of TAFI have been associated with a moderate increase in the risk for venous thromboembolism.7,8 The fibrinolytic system plays a key role in balancing the procoagulant forces of the coagulation cascade to ensure that vascular patency is not sacrificed to maintain vascular continuity (Fig. 21-1). Vascular injury exposes tissue factor to initiate activation of the coagulation cascade through the extrinsic pathway of coagulation. Subsequent activation of the intrinsic pathway leads to amplification of the coagulation response and ultimately hemostatic plug formation. In addition to the negative regulatory response of endogenous anticoagulant proteins such as the protein C/protein S complex and antithrombin, the fibrinolytic system is also activated to dampen and focus clot formation strictly at the site of vascular injury. tPA is released from the surrounding undamaged endothelium in response to thrombin and venous stasis. tPA activates plasminogen poorly in solution but efficiently activates fibrin-bound plasminogen to form plasmin, which initiates clot dissolution. Circulating PAI-1 and α2-antiplasmin efficiently inactivate tPA and plasmin, respectively, in solution, thereby ensuring that systemic activation of fibrinolysis does not occur. Conversely, covalent cross-linking of fibrin monomers and incorporation of α2-antiplasmin into the substance of the clot ensure that clot lysis does not proceed too rapidly before vascular wall repair has progressed sufficiently to prevent delayed rebleeding. Inactivation of fibrin-bound plasmin is greatly slowed in comparison to plasmin free in solution. This mechanism serves to concentrate activation of plasmin on the surface of the clot. Initial fibrinolysis exposes free lysine residues that serve as high-affinity binding sites for plasminogen and increase the sensitivity of plasminogen to activation by tPA. This mechanism serves to accelerate fibrinolysis as it progresses and is negatively regulated by TAFI, which as a carboxypeptidase prunes away these free lysine residues. The interaction of these components ensures focused self-limited clot formation and remodeling that preserves vascular integrity and patency (see Fig. 21-1).
Principles of Thrombolytic Agents
Biology of the Fibrinolytic System
Plasminogen/Plasmin
 For more information on the molecular structure and function of plasminogen, including a structural diagram, please visit the website www.expertconsult.com.
For more information on the molecular structure and function of plasminogen, including a structural diagram, please visit the website www.expertconsult.com.
α2-Antiplasmin
 More information on α2-antiplasmin can be found at www.expertconsult.com.
More information on α2-antiplasmin can be found at www.expertconsult.com.
α2-Macroglobulin
Plasminogen Activator Inhibitors
 More information on the molecular function of PAI-1 can be found at www.expertconsult.com.
More information on the molecular function of PAI-1 can be found at www.expertconsult.com.
Thrombin-Activatable Fibrinolysis Inhibitor
A Model of Regulated Fibrinolysis
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


