Human prion diseases, in common with other neurodegenerative diseases, may be sporadic or inherited and are characterized by the accumulation of cellular proteins accompanied by neuronal death and synaptic loss. Prion diseases are, however, unique in being transmissible. Central to the pathogenesis of all forms of prion disease is the prion protein. This article provides a brief overview of the biology of human prion diseases followed by a more in-depth discussion of the neuropathology of these diseases, including features of neuroradiologic relevance.
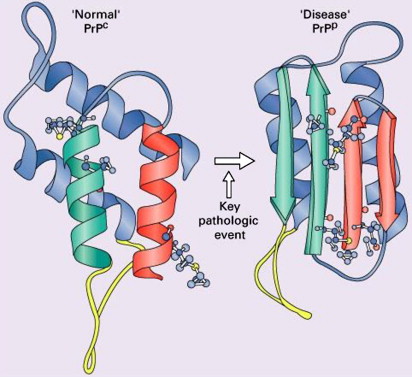
Human prion diseases, in common with other neurodegenerative disease such as Alzheimer’s disease and Parkinson’s disease, are characterized by the deposition of an aberrant form of a constitutively expressed protein in both the sporadic and inherited forms of the disease . The synonymous term “transmissible spongiform encephalopathy,” however, reflects an additional and unique infectious nature indicating acquired forms of disease . The prion diseases occur in a wide range of mammals, including humans . Scrapie, a form of spongiform encephalopathy affecting sheep, has been recognized for more than 200 years, but its cause remained obscure until the 1930s when experimental transmission by contaminated brain was first demonstrated. The transmissibility of human forms of spongiform encephalopathy was proposed after the observation of remarkable similarities in the neuropathology of scrapie and that of kuru, a fatal neurodegenerative disorder showing epidemic spread among the cannibalistic Fore tribe in Papua New-Guinea in the 1950s . Subsequent transmission studies demonstrated the transmissibility of kuru and later of Creutzfeldt-Jakob disease (CJD), a rare human neurodegenerative disease with spongiform pathology, to chimpanzees . Later work elucidating the nature of the agent responsible for the transmission of scrapie favored a new, unique form of protein-related infectivity, but controversy around a “protein only” versus “protein plus virus” (or at least a co-infectious nucleic acid component) persisted. Prusiner introduced the term “prion” to distinguish the proteinaceous infectious particles causing scrapie, CJD, kuru, and familial forms of prion disease from viroids and viruses. The operational definition of the prion as an infectious agent expanded into a structural biologic one according to which prions are proteins that exist in at least two different conformations, one of which can induce the conversion of other prion molecules from one confirmation into the other. This property confers a heritable and self-perpetuating character potentially not requiring cotransmission of nucleic acid . It now is generally accepted that prions consist essentially of an abnormally folded, protease-resistant, beta-sheet rich isoform (abbreviated PrP Sc ) of a normal cellular protein termed “PrP C ” ( Fig. 1 ) .
The prion protein
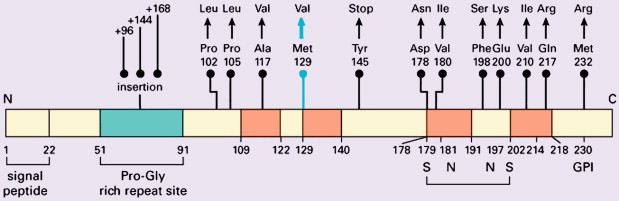
Weismann determined the amino-acid composition and sequence of the prion protein in 1985, and the prion protein gene ( PRN P) was mapped to chromosome 20 shortly thereafter . P RNP is a single-copy gene with three exons, exon 1 coding for PrP, a protein of 253 amino acids ( Fig. 2 ) . PrP C shows a developmentally regulated expression pattern in skeletal muscle, kidney, heart, secondary lymphoid organs, and the central nervous system. In the central nervous system high PrP C expression is found in the synaptic membranes of neurons, and the protein also is expressed in astrocytes . Follicular dendritic cells also exhibit high PrP C expression . The exact functions of PrP c are unknown despite its being a highly conserved protein among a wide range of mammal species. Genetically engineered mice devoid of PrP c show only subtle phenotypes apart from their resistance to prion disease . The mature protein is attached to the cell surface, and it has been suggested that it may function as a signal-transducing molecule . Other suggested functions include protease activity, superoxide dismutase activity given copper-binding sites, facilitation of synaptic transmission, immunoregulation, induction of or protection against apoptosis, and the regulation of neural precursor cell proliferation during developmental and adult mammalian neurogenesis . Participation of PrP C in a heat-shock protein–dependent “danger-sensing” mechanism also has been proposed .
Prion propagation
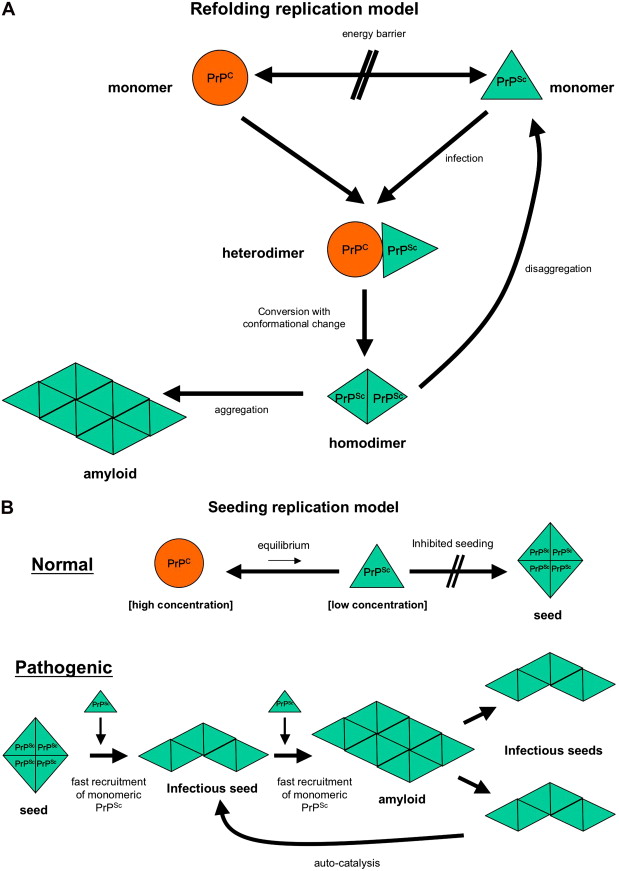
The prion hypothesis proposes that disease progression and infectivity propagate by the recruitment and “autocatalytic” conformational conversion of endogenous PrP C into disease-associated PrP Sc . The presence of PrP C in a given cell type is necessary, but not sufficient, for the replication and propagation of prions . The exact mechanism remains uncertain. The “template-directed refolding hypothesis” (refolding model) proposed by Prusiner holds that an entropic barrier normally prevents conversion of PrP C to PrP Sc . According to this model, monomeric PrP Sc binds with monomeric PrP C to form a heterodimer. This association induces conformational change of the PrP C molecule into a PrP Sc molecule resulting in a homodimer of two PrP Sc molecules. The latter may undergo dissolution into monomeric PrP Sc , allowing a self-propagating cycle of PrP Sc generation, or aggregate into amyloid with consequent tissue deposition ( Fig. 3 A). A more favored theory is the “seeded nucleation hypothesis” (seeding model), which states that PrP Sc and PrP C coexist in equilibrium normally heavily shifted towards PrP C . The extremely low concentrations of PrP Sc under these circumstances inhibit the formation of aggregates (seeds of PrP Sc ). More ordered aggregates of PrP Sc molecules could, however, perform an autocatalytic function with more efficient recruitment of monomeric PrP Sc molecules into highly ordered PrP Sc aggregates (seeds), allowing infectivity with transmission. Such seeds may aggregate further into amyloid with tissue deposition or fragment, allowing the propagation of infectious seeds ( Fig. 3 B). Current experimental evidence favors this mechanism with supportive evidence coming from yeast models of prion replication .
Peripheral prion pathogenesis
Neuroinvasion and the development of prion disease in humans and animals following infection caused by non–central nervous system inoculation of pathogenic prion protein requires the extracerebral tissue/cell expression of non-pathogenic prion protein . PrP-expressing B and T lymphocytes, macrophages, and dendritic cells may facilitate the transport of prions from the peripheral entry site to secondary lymphoid organs . The primary cellular reservoir responsible for prion accumulation and replication before neuroinvasion, however, seem to be the follicular dendritic cells in these lymphoid tissues . Further migration into the nervous system may be facilitated by the sympathetic nervous system, which is primarily responsible for the innervation of lymphoid organs . Prion replication within follicular dendritic cells also has been shown to occur in foci of chronic lymphocytic inflammation outside lymphoid organs . This observation raises the possibility of horizontal spread of prions mediated by secreted body fluids (for example urine and milk) derived from inflamed kidney or mammary tissue. Oral infection in animals is associated with an early increase in prion infectivity in gut-associated lymphoid tissues in the distal ileum . Mobile dendritic cells may facilitate the transport of infected prion proteins between the gut lumen and lymphoid replication sites where it also may be involved in the eventual transport of prions from these sites of replication to peripheral nerves within the lymphoid organs.
Prion strains
According to the “prion-only” hypothesis, distinct versions of prion disease within the same mammalian species are determined (at least in part) by prion protein strains. Strain variation is a function of various distinct pathologic conformations, each of which can impart it own conformation onto PrP C . Structural variation in PrP Sc potentially contributing to conformational variation and to pathogenetic expression may be explained by mutations in the coding region of the PRNP gene in inherited forms of prion disease . Furthermore, the PRNP gene is polymorphic at codon 129, encoding for either valine (V) or methionine (M) . Homozygosity for methionine confers greater susceptibility to sporadic CJD (sCJD) , and the codon 129 polymorphism may influence the clinicopathologic phenotype of prion diseases profoundly. Isotypes of PrP Sc may be identified using Western blot analysis after limited proteolysis, causing differing degrees of N-terminal truncation and resulting in nonglycosylated fragments of differing molecular weight and electrophoretic mobility. Further discrimination of type 2 PrP Sc as type 2A and type 2B may be made on the basis of the glycosylation pattern at two potential sites in the protein .
Classification of human prion diseases
The classification of human prion diseases recognizes idiopathic (sCJD), inherited, and acquired forms of disease . The first verified cases of what later became to be known as “Creutzfeldt-Jakob” disease were reported in 1921 by Jakob. This report included familial cases later shown to be caused by a mutation in the PRNP gene. The spectrum of inherited human prion diseases now includes different forms of familial CJD, classical and variant forms of Gerstmann-Sträussler-Scheinker syndrome (GSS), and fatal familial insomnia (FFI). These conditions have been linked to an ever-increasing number of pathogenic mutations and insertions in the PRNP gene . Kuru is the first identified acquired form of human prion disease. The first iatrogenic acquired case of CJD was reported in 1974 in a corneal transplant recipient . Iatrogenic CJD related to dura mater grafts and human-derived pituitary hormone treatment has been described subsequently . A new variant form of CJD (vCJD) was reported in 1996 in the United Kingdom . Epidemiologic evidence has linked this disorder to the epidemic of bovine spongiform encephalopathy in the United Kingdom since 1986, drawing considerable public attention to prion diseases. Biochemical and experimental transmission studies have provided compelling evidence in support of vCJD as the first example of a zoonotic form of prion disease in humans . The identification of cases of CJD acquired as a result of blood transfusions is a worrying recent development . The most common human prion disease, however, is still idiopathic sCJD (with an incidence of approximately 1 case per million population per year), which is not associated with any known mutations in the PRNP gene or environmental risk factors. The clinical features of the human prion diseases are summarized in Table 1 .
| Subtype | Age at onset in years (range) | Disease duration (range) | Main clinical features | Cerebrospinal fluid 14-3-3 protein | EEG | Codon 129 | PRNP mutation |
|---|---|---|---|---|---|---|---|
| Idiopathic CJD | |||||||
| Sporadic CJD | Mean 65 (14–92) | Median 4.5 mo mean 8 mo (2 wk–35 mo) | Progressive dementia, myoclonus, visual cerebellar disturbances, pyramidal–extrapyramidal signs, akinetic mutism | Positive in 93% | PSWC (66%–90%) | MM 70% MV 14% VV 16% | Not demonstrable |
| sFI | Mean 60.3 | 14 mo | Autonomic dysfunction bulbar and gait motor signs | Negative | Nonspecific alterations | MM | Not demonstrable |
| Inherited CJD | |||||||
| Familial CJD | 50–60 (26–81) | 6 mo (2–41 months) | Almost similar to sCJD MM1, but haplotype-related variability | Positive in more than 90% | PSWC 70% | More than 25 disease-associated mutations | |
| GSS | 50–60 (2nd–8th decade) | 5–6 y (3 mo–13 y) | Progressive ataxia, incoordination, dysarthria, pyramidal/extrapyramidal signs, late dementia | Usually negative | Nonspecific alterations | M (on the mutated allele) | P102L (plus several less common mutations) |
| FFI | 50 (20–63) | 13–15 mo (6–42 mo) | Insomnia, autonomic dysfunction, bulbar and gait motor symptoms | Negative | Nonspecific alterations | M (on the mutated allele) | D1789 |
| Variable phenotypes | Progressive dementia in most, myoclonus, cerebellar and extrapyramidal signs | Nonspecific alterations | Octapeptide repeat region inserts | ||||
| Acquired Creutzfeldt-Jakob disease | |||||||
| Variant CJD | 26 (12–74) | 14 mo (6–39 mo) | Early psychiatric symptoms (depression, anxiety, delusions, withdrawn), persistent painful sensory symptoms, ataxia, myoclonus/chorea/dystonia, later dementia | 50% | Nonspecific alterations, No PSWC | MM 99% | Not demonstrable |
| Iatrogenic CJD | 37.7 (dural grafts); 9.6 (growth hormone recipients) | Similar to sCJD (incubation period 1–30 y: 12–28 mo with cerebral inoculation and 5–30 y for growth hormone recipients) | Clinical symptoms similar to sCJD, but usually cerebellar onset with peripheral infections and usually dementing onset with intracerebral infections. | 77% | Similar to sCJD | MM 57% MV 20% VV 23% | Not demonstrable |
| Kuru | 5–60 | 12 mo | Progressive ataxia, tremors, emotional liability, choreoathetotic involuntary movements, dementia not a dominant feature | MM genotype preferentially affected but MV and VV genotypes also present | No demonstrable | ||
Murine studies have supported the notion of latent or subclinical infection . These studies and evidence of incomplete penetrance in older people carrying PRNP mutations raises the possibility of natural chronic asymptomatic infection/disease that might maintain subclinical reservoirs.
Classification of human prion diseases
The classification of human prion diseases recognizes idiopathic (sCJD), inherited, and acquired forms of disease . The first verified cases of what later became to be known as “Creutzfeldt-Jakob” disease were reported in 1921 by Jakob. This report included familial cases later shown to be caused by a mutation in the PRNP gene. The spectrum of inherited human prion diseases now includes different forms of familial CJD, classical and variant forms of Gerstmann-Sträussler-Scheinker syndrome (GSS), and fatal familial insomnia (FFI). These conditions have been linked to an ever-increasing number of pathogenic mutations and insertions in the PRNP gene . Kuru is the first identified acquired form of human prion disease. The first iatrogenic acquired case of CJD was reported in 1974 in a corneal transplant recipient . Iatrogenic CJD related to dura mater grafts and human-derived pituitary hormone treatment has been described subsequently . A new variant form of CJD (vCJD) was reported in 1996 in the United Kingdom . Epidemiologic evidence has linked this disorder to the epidemic of bovine spongiform encephalopathy in the United Kingdom since 1986, drawing considerable public attention to prion diseases. Biochemical and experimental transmission studies have provided compelling evidence in support of vCJD as the first example of a zoonotic form of prion disease in humans . The identification of cases of CJD acquired as a result of blood transfusions is a worrying recent development . The most common human prion disease, however, is still idiopathic sCJD (with an incidence of approximately 1 case per million population per year), which is not associated with any known mutations in the PRNP gene or environmental risk factors. The clinical features of the human prion diseases are summarized in Table 1 .
| Subtype | Age at onset in years (range) | Disease duration (range) | Main clinical features | Cerebrospinal fluid 14-3-3 protein | EEG | Codon 129 | PRNP mutation |
|---|---|---|---|---|---|---|---|
| Idiopathic CJD | |||||||
| Sporadic CJD | Mean 65 (14–92) | Median 4.5 mo mean 8 mo (2 wk–35 mo) | Progressive dementia, myoclonus, visual cerebellar disturbances, pyramidal–extrapyramidal signs, akinetic mutism | Positive in 93% | PSWC (66%–90%) | MM 70% MV 14% VV 16% | Not demonstrable |
| sFI | Mean 60.3 | 14 mo | Autonomic dysfunction bulbar and gait motor signs | Negative | Nonspecific alterations | MM | Not demonstrable |
| Inherited CJD | |||||||
| Familial CJD | 50–60 (26–81) | 6 mo (2–41 months) | Almost similar to sCJD MM1, but haplotype-related variability | Positive in more than 90% | PSWC 70% | More than 25 disease-associated mutations | |
| GSS | 50–60 (2nd–8th decade) | 5–6 y (3 mo–13 y) | Progressive ataxia, incoordination, dysarthria, pyramidal/extrapyramidal signs, late dementia | Usually negative | Nonspecific alterations | M (on the mutated allele) | P102L (plus several less common mutations) |
| FFI | 50 (20–63) | 13–15 mo (6–42 mo) | Insomnia, autonomic dysfunction, bulbar and gait motor symptoms | Negative | Nonspecific alterations | M (on the mutated allele) | D1789 |
| Variable phenotypes | Progressive dementia in most, myoclonus, cerebellar and extrapyramidal signs | Nonspecific alterations | Octapeptide repeat region inserts | ||||
| Acquired Creutzfeldt-Jakob disease | |||||||
| Variant CJD | 26 (12–74) | 14 mo (6–39 mo) | Early psychiatric symptoms (depression, anxiety, delusions, withdrawn), persistent painful sensory symptoms, ataxia, myoclonus/chorea/dystonia, later dementia | 50% | Nonspecific alterations, No PSWC | MM 99% | Not demonstrable |
| Iatrogenic CJD | 37.7 (dural grafts); 9.6 (growth hormone recipients) | Similar to sCJD (incubation period 1–30 y: 12–28 mo with cerebral inoculation and 5–30 y for growth hormone recipients) | Clinical symptoms similar to sCJD, but usually cerebellar onset with peripheral infections and usually dementing onset with intracerebral infections. | 77% | Similar to sCJD | MM 57% MV 20% VV 23% | Not demonstrable |
| Kuru | 5–60 | 12 mo | Progressive ataxia, tremors, emotional liability, choreoathetotic involuntary movements, dementia not a dominant feature | MM genotype preferentially affected but MV and VV genotypes also present | No demonstrable | ||
Murine studies have supported the notion of latent or subclinical infection . These studies and evidence of incomplete penetrance in older people carrying PRNP mutations raises the possibility of natural chronic asymptomatic infection/disease that might maintain subclinical reservoirs.
Neuropathology of human prion diseases—general features
Gross pathology
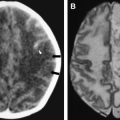
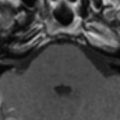
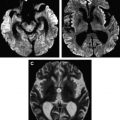
In cases of sporadic, iatrogenic, and familial CJD, the gross appearance of the brain may vary from normal to showing variable degrees of cerebral cortical, striatal, and/or cerebellar atrophy. Some degree of diffuse or focal cerebral atrophy usually is apparent. Occipital, striatal, thalamic, and cerebellar variants have been described based on macroscopic appearances ( Fig. 4 A, C) . Severe atrophy may correlate with longstanding disease, and the brain weight may be as low as 850 g . In general, loss of brain substance is confined to gray matter with relative sparing of white matter. In cases with severe atrophy, ventricular enlargement is marked with atrophy of the caudate nucleus and thalamus. The hippocampal formation usually is well preserved even in cases of severe brain atrophy, in contrast to other degenerative dementias, including Alzheimer’s disease . Most cases of vCJD show no macroscopic abnormalities. Long disease duration (in excess of 19 months), however, may manifest cerebellar atrophy preferentially affecting the vermis. This atrophy is accompanied by mild cerebral cortical atrophy, most conspicuous in the primary visual cortex. The hippocampi, subcortical white matter, basal ganglia, and thalami usually show normal macroscopic appearances . Significant cerebral and cerebellar atrophy also may become apparent only with long disease duration (in excess of 30 months) in patients who have familial and sporadic forms of fatal insomnia .
Histopathology
The spectrum of pathologic alterations described in the human prion diseases shows individual variation in specificity, intensity, and distribution. These variations allow the recognition of characteristic but (given overlapping pathology) not always exclusive phenotypes. The neuropathology is subject to disease subtype (sporadic, familial, acquired), the nature of the PRNP gene defect (if familial), the codon 129 haplotype (MM, MV, or VV), the PrP Sc strain type, and the duration of illness. The pathologic changes present in cases coming to biopsy or autopsy usually are moderate to marked, and the absence of pathologic changes readily demonstrated by routine histologic techniques is very unusual. The pathology in a single specimen may be distinctive enough to allow a definitive diagnosis, but diagnosis cannot be guaranteed, because of geographic variability. The major pathologic features are discussed in the following sections.
Spongiform change
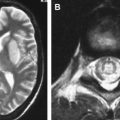
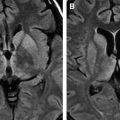
Spongiform change or degeneration is relatively specific to the prion diseases and usually is present. “Spongiform change” refers to vacuolation of the neuropil of the cerebral cortex, the subcortical gray matter, and the cerebellar molecular layer ( Fig. 5 A, C, D) . It is present in most cases of sporadic, iatrogenic, and familial CJD, regardless of the clinical presentation, but may vary considerably from region to region, even within the same section . In cases with equivocal or no spongiform change, PrP immunohistochemistry or Western blot analysis is required to make a definitive diagnosis . The spongiform change can be distributed diffusely in all cortical layers or may have a pseudolaminar appearance primarily affecting the deep cortical layers. The identification of vacuoles within the neuropil away from cell bodies and blood vessels is helpful diagnostically, because vacuolation in association with the latter structures is less specific. The vacuoles typically are round to oval, relatively small (mainly 5–20 μm but reaching 50 μm) in diameter, and usually are distributed quite evenly. The vacuoles may become confluent, resulting in larger, more irregular vacuoles with a size of up to 200 μm, which may distort the cortical cytoarchitecture substantially. The cerebellum often shows confluent spongiform change or exhibits widespread microvacuolar change with smaller vacuoles, 20 to 50 μm in diameter, in the molecular layer. Spongiform change is almost always present in the head of the caudate nucleus in cases of sporadic disease but usually is minimal to absent in brain stem and spinal cord . Ammon’s horn and the dentate gyrus of the hippocampus usually are spared, but the subiculum may be affected. The smaller, more characteristic vacuoles are shown by electron microscopy to consist of focal swellings of neuritic processes (axons and dendrites) and synapses, representing intracellular vacuolation ( Fig. 5 E) . Affected neuritic processes show loss of internal organelles and the formation of lacy, abnormal membranous structures. A membrane may enclose small vacuoles. Secondary chamber formation may be seen within the vacuoles. Larger vacuoles usually are incompletely enclosed or lack any discernable surrounding membrane. Curled membranous fragments and amorphous material may be found in the areas of vacuolation. Swelling of organelles such as the endoplastic reticulum and Golgi apparatus may be present . Peculiar tubulovesicular structures have been described as a specific ultrastructural marker of CJD . Vacuolation of nerve cell bodies in human prion diseases has been described (usually involving larger neurones) but is unusual (apart from kuru), in contrast to transmissible spongiform encephalopathies of animals such as scrapie . The relative disappearance of spongiform change with progressive neuronal loss is consistent with the hypothesis that vacuolation is confined mostly to nerve cell processes . Although white matter pathology is thought to be mainly secondary to neuronal loss, spongiform change also may be evident here . A vacuolar myelopathy may be found in some cases of CJD. Ultrastructurally the vacuoles were found within the myelin sheaths but occasionally may be intra-axonal also. Extensive white matter degeneration is present in the panencephalopathic form of CJD, which is seen more frequently in Japan .
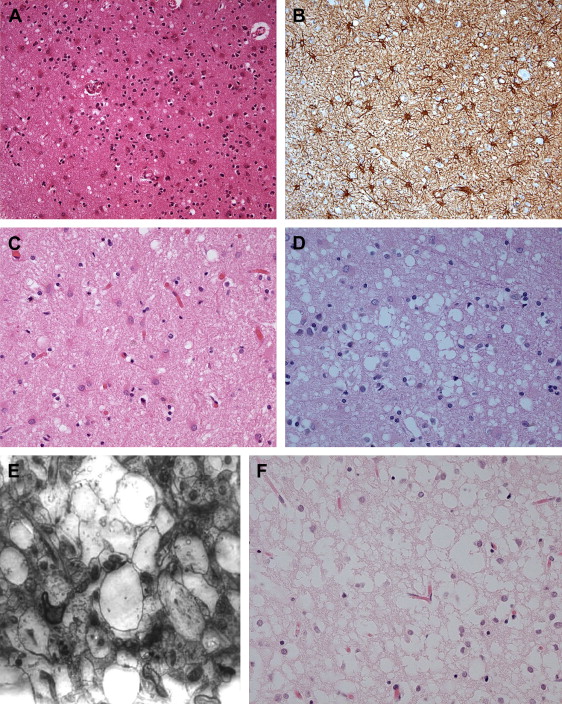
With long disease duration the cortex may be reduced to a distorted rim of gliotic tissue containing a few remaining neurons against a background of extensive coarse vacuolation (100–200 μm) surrounded by a dense meshwork of astrocytic gliosis ( Fig. 5 F) . This pattern of cortical injury is referred to as “status spongiosis,” a nonspecific finding common to several neurodegenerative diseases in an advanced stage of development. Microvacuolation similar to that seen in prion diseases may be found in other neurodegenerative diseases such as fronto-temporal lobar dementia and dementia with Lewy bodies as well as in some metabolic encephalopathies and acute hypoxic/ischemic encephalopathy. The distribution of the microvacuolar change and the presence and absence of other relevant pathologic findings, however, allows distinction from these conditions. The absence of spongiform change was shown to be an almost 100% predictive indicator of transmissibility of sporadic and familial human CJD to laboratory animals .
Neuronal loss
Most cases show significant neuronal loss, but the pattern of loss may be variable. The loss usually is exaggerated in cortical layers III to V and in focal regions of the caudate nucleus and thalamus . In the cerebellum there may be irregular loss of neurones in the granular cell and Purkinje cell populations. With status spongiosis, neuronal loss is extensive, causing a loss of lamination. Scattered shrunken neuronal rims may be seen admixed with apparently unaffected cells. Some residual neurones may show ballooning related to the accumulation to neurofilament protein . Experimental evidence suggests that nerve cell death is a late event . Neuronal loss in affected cortical and subcortical regions seems to follow an apoptotic pathway. The latter may correlate with microglial activity and axonal damage rather than the local deposition of PrP Sc . Selective vulnerability of a subset of inhibitory GABAergic neurons has been described .
Astrocytic gliosis
Reactive astrocytosis in CJD seems more intense that would be predicted by the degree of nerve cell loss (see Fig. 5 A, B) . The degree of reactive astrocytosis in CJD, however, was shown to be directly proportional to the degree of neuronal loss . The intense astrocytic reaction in prion disease may be explained by stimulation of astrocyte proliferation by fractions of the prion protein .
Microgliosis
Microglial hypertrophy and hyperplasia show a widespread distribution in prion disease. Ultrastructural and immunohistochemical studies of both human and animal prion diseases have demonstrated the intimate involvement of microglial cells in PrP plaque formation. This finding suggests a role in the amyloidogenic processing of PrP. Unlike other infectious disorders, prion diseases are associated with only scant, if any, lymphocytic-mediated inflammatory activity or the accumulation of foamy macrophages .
Synaptic loss
Ultrastructural studies have shown that axons and dendrites are damaged early during the disease course. Synaptic loss of approximately 20% has been demonstrated in atypical cases lacking obvious pathology at a light microscopic level. In cases with obvious spongiform change, a 30% reduction in synaptic density has been estimated .
Accumulation of PrP
The accumulation of PrP Sc can be detected by immunohistochemistry and allows a definitive diagnosis of prion disease. PrP accumulation seems to precede the other pathologic changes . Several patterns of PrP Sc accumulation have been described. The occurrence and/or prevalence of these patterns, again, are related to the PrP Sc type, codon 129 polymorphism, and brain region.
- •
The diffuse synaptic pattern comprises usually abundant, punctate immunolabeling, occasionally accompanied by coarser and bigger deposits throughout the neuropil ( Fig. 6 A). In cases with minimal cerebral cortical deposition, cerebellar involvement remains obvious . The granular layer most frequently is affected in the cerebellar cortex, with coarse granules with additional but variable fine stippling of the molecular layer.