Pulmonary arterial hypertension (PAH) is defined as a mean pulmonary arterial pressure of greater than 25 mm Hg at rest or 30 mm Hg with exercise, with an elevated pulmonary vascular resistance (PVR) greater than 3 Wood units. Although pulmonary pressures may be elevated in many conditions, PAH fundamentally requires changes in the pulmonary vasculature. Mean pressures of 26 to 40 mm Hg are considered mild; 41 to 55 mm Hg, moderate; and greater than 55 mm Hg, severe.
Although the inciting factor may be from any number of conditions, PAH is due to progressive increase of PVR, leading to increased right ventricle (RV) pressure load. As PAH progresses, the RV is unable to compensate, leading initially to exertional symptoms (usually dyspnea) but eventually to florid right heart failure, cardiogenic shock, and death.
The diagnosis of PAH continues to be a challenge, predominantly because of nonspecific initial symptoms. Although pathologic findings are definitive for the diagnosis, routine biopsies are not performed, and therefore noninvasive imaging is essential for diagnosis. Largely, this is achieved via echocardiography, but this is not ordered unless clinical suspicion of PAH is high. Other imaging modalities are essential in increasing or decreasing the index of suspicion. Once PAH is suspected, imaging is also essential for identifying the underlying etiology and comorbid conditions. However, evaluation of the degree of pulmonary pressure elevation, changes in PVR, and any contribution of left-sided heart disease is best done by hemodynamic catheterization (right heart catheterization), especially if considering PAH-specific therapy.
Etiology and Classification
PAH can be usefully classified according to the predominant site of pathologic insult, into precapillary and postcapillary causes ( Box 53.1 ). Precapillary causes include small-vessel vasculopathy affecting the arterioles, thrombotic or embolic disease, proximal vascular obstruction by tumor or vasculitis, congenital heart disease (CHD), and pleuroparenchymal disease. Postcapillary causes of pulmonary hypertension include causes of chronically raised pulmonary venous pressure, such as left ventricular failure, valvular heart disease, atrial myxoma, mediastinal fibrosis, and, rarely, congenital venous stenosis, as well as predominantly postcapillary vasculopathies, such as pulmonary venoocclusive disease, and pulmonary capillary hemangiomatosis (PCH).
Precapillary Hypertension
Small-vessel disease
Idiopathic pulmonary arterial hypertension
Portopulmonary hypertension
Anorexigen abuse
Connective tissue associated
Human immunodeficiency virus infection
Pulmonary arterial obstruction and stenosis
Chronic thromboembolic disease
Nonthrombotic emboli (metastatic neoplasm, parasites, talc)
Sickle cell disease
Increased blood flow
Congenital heart disease or shunts
Pleuropulmonary disease
Chronic obstructive pulmonary disease
Interstitial lung disease
Bronchiectasis, cystic fibrosis
Chest wall deformities
Alveolar hypoventilation
Neuromuscular disease
Obesity
Obstructive sleep apnea
Postcapillary Hypertension
Pulmonary venoocclusive disease
Pulmonary capillary hemangiomatosis
Left-sided heart failure
Mitral valve disease
Myxoma
Mediastinal fibrosis
Congenital venous stenosis
Anomalous pulmonary venous connections
A more clinically based classification of pulmonary hypertension has been developed and refined, aiming to individualize different categories sharing similarities in pathophysiologic mechanisms, clinical presentation, and therapeutic options. These two classifications inevitably bear many similarities. The World Health Organization meeting in 1973 was the first to endorse standardized nomenclature. Since then, the clinical classification has been reclassified multiple times. The most recent classification system was devised in 2009 in Dana Point, California, and was essentially maintained, with some minor adjustments, at the Fifth World Symposium on Pulmonary Hypertension in Nice, France ( Box 53.2 ). The last three iterations have largely been minor reclassifications to try to better align the pathophysiology of PAH in different conditions.
Group 1. Pulmonary arterial hypertension
- 1.1.
Idiopathic
- 1.2.
Heritable
- 1.3.
Drug and toxin induced
- 1.4.
Associated with
- 1.4.1.
Collagen vascular disease
- 1.4.2.
Human immunodeficiency virus infection
- 1.4.3.
Portal hypertension
- 1.4.4.
Congenital heart disease
- 1.4.5.
Schistosomiasis
- 1.4.6.
Chronic hemolytic anemia
- 1.4.1.
- 1.5.
Pulmonary venoocclusive disease or pulmonary capillary hemangiomatosis
- 1.6.
Persistent pulmonary hypertension of the newborn
- 1.1.
Group 2. Pulmonary hypertension with left-sided heart disease
- 2.1.
Systolic dysfunction
- 2.2.
Diastolic dysfunction
- 2.3.
Left-sided valvular heart disease
- 2.4.
Congenital/acquired left ventricular inflow or outflow obstruction or congenital cardiomyopathy
- 2.1.
Group 3. Pulmonary hypertension associated with lung disease or hypoxemia
- 3.1.
Chronic obstructive pulmonary disease
- 3.2.
Interstitial lung disease
- 3.3.
Other pulmonary diseases with mixed restrictive and obstructive pattern
- 3.4.
Sleep-disordered breathing
- 3.5.
Alveolar hypoventilation disorders
- 3.6.
Chronic exposure to high altitude
- 3.7.
Developmental abnormalities
- 3.1.
Group 4. Pulmonary hypertension resulting from chronic thrombotic or embolic disease
Group 5. Pulmonary hypertension with unclear, multifactorial mechanisms
- 5.1.
Hematologic disorders: myeloproliferative disorders, splenectomy
- 5.2.
Systemic disorders: sarcoidosis, Langerhans cell histiocytosis, lymphangioleiomyomatosis, neurofibromatosis, vasculitis
- 5.3.
Metabolic disorders: glycogen storage disease, Gaucher disease, thyroid disorders
- 5.4.
Others: compression of pulmonary vessels, fibrosing mediastinitis, chronic renal failure on dialysis, segmental pulmonary hypertension
- 5.1.
Group 1 PAH is a varied group of underlying conditions that result in increased pulmonary vascular tone from intrinsic issues in the pulmonary vasomotor tone or a secondary increase in PVR from an increase in pulmonary vascular flow. Group 2 is pulmonary hypertension related to left-sided heart disease, whereas pulmonary hypertension associated with pulmonary parenchymal disease and/or hypoxemia is group 3. Chronic thrombotic or embolic diseases comprise group 4, and group 5 is a heterogeneous group with multifactorial or unclear mechanisms (see Box 53.2 ).
Some important developments in the understanding of group 1 PAH over the years include recognition of specific genetic mutations associated with PAH, as well as drug- and toxin-mediated PAH. Familial PAH is an autosomal-dominant condition with incomplete penetrance, caused in 50% of cases by mutations in the bone morphogenetic protein receptor type II, and it is clinically and radiologically indistinguishable from idiopathic PAH. The most recognized drugs associated with PAH are the anorexigenic drugs, namely, aminorex fumarate, fenfluramine, and dexfenfluramine. Cocaine, amphetamine, and chemotherapeutic agents have also been implicated.
Group 1 PAH may also develop from a sustained congenital left-to-right shunt. This includes both high-pressure shunts, such as truncus arteriosus, ventricular septal defects, and patent ductus arteriosus (PDA), and low-pressure high-flow shunts, such as atrial septal defects and partial anomalous pulmonary venous return. Other conditions, such as chronic renal failure, portopulmonary hypertension, and thyrotoxicosis, which increase pulmonary blood flow, can also result in pulmonary hypertension.
Initially, the presence of a shunt exposes the pulmonary circulation to high flow, preventing it from adapting normally to extrauterine life. Ongoing high flow during extended periods results in a progressive pulmonary vasculopathy with associated in situ thrombosis. The position and size of the cardiac defect and the magnitude of the shunt govern the time taken for these changes to occur. Left uncorrected, rising pulmonary arterial pressures lead to bidirectional and then reversed flow across the shunt (Eisenmenger syndrome). Even after apparently effective corrective surgery, patients may go on to develop significant pulmonary hypertension.
Pulmonary hypertension occurs in a wide variety of collagen vascular diseases and confers a worse prognosis. Incidences vary among the diseases but can be as high as 12% in patients with the limited form of scleroderma. Pulmonary hypertension is the cause of up to 50% of disease-related deaths in patients with limited scleroderma. Isolated pulmonary hypertension is less common in diffuse scleroderma; when it is present, it is often seen in patients with the nucleolar antibody anti-U3 ribonucleoprotein. In mixed connective tissue disease (CTD), one long-term follow-up study found that pulmonary hypertension was the most common cause of death, occurring in 38% of patients. Although elevated PA pressures can be seen with any CTD, it is less frequently seen in systemic lupus erythematosus, rheumatoid arthritis (RA), and polymyositis. In RA, pulmonary hypertension is more frequently a late manifestation of rheumatoid interstitial lung disease than of vasculopathic changes. Systemic lupus erythematosus can be complicated by a hypercoagulable state; therefore chronic thromboembolic pulmonary hypertension (CTEPH) should always be considered an alternative cause of pulmonary hypertension in these patients.
Pulmonary hypertension is thought to affect 0.1% of the human immunodeficiency virus (HIV)-infected population annually, representing a relative risk of 500, compared with an HIV-negative individual. Pulmonary thromboembolic disease represents another cause of pulmonary hypertension in patients with chronic HIV infection, being present in 3% of patients with pulmonary hypertension and HIV infection or acquired immune deficiency syndrome.
Pulmonary venoocclusive disease (PVOD) and PCH are considered together because they share several common features, particularly in relation to their management. Although an arterial component is usually present, both diseases predominantly affect the venules immediately downstream from the capillary bed. The venous occlusion in PCH is due to an abnormal uncontrolled proliferation of capillaries that infiltrate the interstitium, the vessels, and, less commonly, the airways. In both PVOD and PCH, venous occlusion results in increased transcapillary hydrostatic pressure, with consequent focal areas of edema and hemorrhage. There is limited epidemiologic data available on both diseases. PVOD is rare, with an incidence of 0.1 to 0.3 per million, although up to 5% to 10% of idiopathic PAH patients have been found on pathology to have PVOD. PCH is rarer still. Unlike idiopathic PAH, one-third of PVOD cases occur in children, with equal sex distribution; there is a (2 : 1) male predominance in adult patients.
The etiology of both diseases is unknown and may be multifactorial. Cytotoxic drugs, including bleomycin, herbal “bush” tea, bone marrow transplantation, and thoracic radiotherapy have all been implicated in PVOD, although the majority of cases are currently considered idiopathic. Clinically, the triad of severe PAH, radiographic evidence of pulmonary edema, and normal pulmonary artery occlusion pressure is considered to be diagnostic of PVOD. In conjunction, these findings can obviate the need for tissue diagnosis. However, many patients with PVOD do not have this triad. As these diseases are often difficult to distinguish from idiopathic pulmonary hypertension and from interstitial lung disease, radiologic investigations, particularly computed tomography (CT), have an important role to play in diagnosis.
Many chronic lung conditions can lead to pulmonary hypertension through an increase in PVR. This is partly due to destruction of the vascular bed and partly due to vascular remodeling in response to chronic alveolar hypoxia. Although the resulting pulmonary hypertension is often mild and associated with a preserved cardiac output, its presence worsens prognosis. Approximately 90% of patients with severe chronic obstructive pulmonary disease will have pulmonary hypertension.
In CTEPH thrombi from untreated or recurrent acute emboli organize and become incorporated into the wall of the pulmonary arteries. Thrombi in smaller vessels often recanalize, forming a trabecular mesh. This process, which may occur in repeated cycles, leaves endothelialized residua that obstruct or narrow pulmonary arteries, resulting in progressive pulmonary hypertension, hypoxemia, and right-sided heart failure. The true prevalence of CTEPH is unknown as many cases may remain undiagnosed. CTEPH may complicate up to 3.8% of cases of acute pulmonary embolism and carries a poor prognosis if it is left untreated.
The importance of aggressively identifying etiology cannot be understated. In particular, PAH-specific vasodilator therapy has been indicated in group 1 (excluding PVOD and PCH). Vasodilator therapy has been studied inconsistently in group 3 (PAH associated with parenchymal lung disease) or group 4 (CTEPH), although it is likely still efficacious. Postcapillary causes of PAH (PVOD, PCH, and group 2 PAH) do not typically benefit from pulmonary vasodilator therapy. Furthermore, the approach to transplantation is varied based on the underlying etiology (single-lung vs. double-lung transplantation vs. multiorgan transplantation).
Epidemiology
Given the challenges in recognition and diagnosis, the true incidence and prevalence have been difficult to capture. There are no large international registries, and national registries have some variance. In addition, most of the registry data has been limited to group 1 PAH, and the epidemiology of other PAH groups is still not entirely clear. However, group 1 PAH still remains a relatively rare disease with an annual incidence of 1.1 to 3.6 per million population and a prevalence of 6.6 to 52 per million population. There is a female preponderance (anywhere from 60%–80%) that has been fairly consistent over time. Although early registries suggested this to be a disease of the young, the median age of more recent registries seems to range from 50 to 65 years of age.
Idiopathic PAH in these registries represents anywhere from 39% to 60% of all group 1 PAH patients, with anywhere from 11% to 43% being PAH associated with CHD and 15% to 30% PAH associated with CTD. Familial or heritable PAH is an increasingly recognized etiology for group 1 PAH; however, idiopathic cases outnumber familial cases of pulmonary hypertension by more than 10 : 1.
Pathophysiology
At birth the pulmonary arterial circulation is histologically similar to the systemic circulation. However, as the newborn adapts to extrauterine life, the pulmonary arterial circulation rapidly develops into a high-flow low-pressure system. Increase in vascular compliance allows the pulmonary circulation to accommodate systemic cardiac outputs with little rise in pulmonary arterial pressure.
Pulmonary arteries more than 1 mm in diameter have walls that consist of numerous parallel elastic laminae. These “elastic” arteries act predominantly as conductance vessels and contribute little to resistance. As the vessel diameter narrows to between 1 mm and 100 µm, the elastic component is replaced by smooth muscle. The precapillary “muscular” arteries contribute significantly to PVR and are, in health, the site of the greatest pressure drop within the circulation. In general, the muscular arteries or resistance vessels are also the site most affected in disease, as occurs in idiopathic PAH.
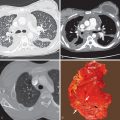
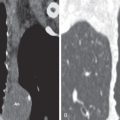
PAH, regardless of cause, results in dilation of the large elastic pulmonary arteries and may result in pulmonary arterial atherosclerosis. Other common findings include intimal fibrosis of elastic and large muscular arteries and thickening of the media of small muscular arteries. In addition to muscle hypertrophy-hyperplasia, several abnormalities are often present in small- to medium-sized muscular arteries that together characterize plexogenic pulmonary arteriopathy. These abnormalities include cellular intimal proliferation and fibrosis, plexiform lesions, fibrinoid “necrosis,” and vasculitis ( Fig. 53.1 ). Plexiform lesions are seen in small supernumerary arteries a short distance beyond their origin from the parent vessel. The lesion consists of a localized focus of vascular dilation associated with an intraluminal plexus of slit-like vascular channels (see Fig. 53.1 ). This plexogenic arteriopathy is the hallmark of idiopathic PAH and PAH associated with congenital systemic-pulmonary shunts (most commonly CHD). The histologic findings of pulmonary hypertension resulting from chronic thromboembolism include thrombi in various stages of organization in the large and small pulmonary arteries. The most common findings in small pulmonary vessels are medial hypertrophy and intimal fibrosis, which may be eccentric, concentric, or in a colander pattern consistent with recanalized thrombus ( Fig. 53.2 ). Venoocclusive disease is characterized by stenosis or obliteration of the lumina of small pulmonary veins and venules by intimal fibrous tissue ( Fig. 53.3 ). Other common findings are engorged venules, dilated lymphatics, and septal thickening (see Fig. 53.3 ). Histologic evidence of PAH is usually present, but changes of plexogenic arteriopathy are absent. The main histologic abnormality in PCH consists of patchy interstitial proliferation of thin-walled blood vessels the size of capillaries. The vessels appear to invade the walls of pulmonary veins and to a lesser extent the pulmonary arteries. The venular infiltration is often accompanied by intimal fibrosis, which may lead to stenosis.



Clinical Presentation and Outcomes
Pulmonary hypertension is often difficult to diagnose because it manifests insidiously with nonspecific clinical findings. Patients initially complain of exertional breathlessness, representing right-sided cardiac insufficiency in the face of an increased workload. As disease progresses, exercise tolerance steadily deteriorates, and exertional syncope or chest pain may develop. Once the right ventricle fails to sustain an appropriate cardiac output at rest, patients develop ongoing symptoms of overt right-sided heart failure, including fatigue, loss of appetite, ascites, and pedal edema. Hence, not uncommonly, pulmonary hypertension can be misdiagnosed as asthma, hyperventilation syndrome, or lack of fitness. The delay in diagnosis compounded by the fact that the pulmonary circulation has an extensive reserve means that by the time the diagnosis is made, much of the circulation has already been compromised.
Prognosis in PAH is still poor long term but has improved over time. National Institutes of Health registry data published in 1991 estimated median survival of 2.8 years for idiopathic PAH, with 1-year, 3-year, and 5-year survival rates of 68%, 48%, and 34%, respectively. More recent data from the REVEAL Registry (Registry to Evaluate Early and Long-Term PAH Disease Management) shows 1-year, 3-year, and 5-year survival rates of 85%, 68%, and 57%, respectively. Prognosis does vary based on etiology, with PAH associated with CHD or drugs and toxins having a better long-term prognosis and PAH associated with portopulmonary hypertension and CTD having a worse long-term prognosis, compared with idiopathic PAH, heritable PAH, or PAH associated with HIV (all with similar prognosis). The prognosis in severe portopulmonary hypertension (coexisting portal hypertension and PAH) is poor, with median survival of 6 months and 5-year survival of 10% in one series, compared with a 5-year survival of 50% for patients with hepatopulmonary syndrome (hypoxemia caused by pulmonary vasodilation and shunting in the setting of liver disease). There are also risk stratification tools proposed by guideline committees that include clinical features (progression of symptoms, syncope), functional parameters (New York Heart Association classification, 6-minute walk distance), and hemodynamic parameters and imaging findings. Specifically, RA area greater than 26 cm 2 and presence of pericardial effusion are considered a high risk for early mortality (1-year mortality >10%).
Synopsis of Treatment Options
Originally, treatment for PAH was limited to anticoagulation, calcium channel blockers, oxygen, and diuretics for symptomatic relief. This is still often the guideline-directed medical therapy for patients who show pulmonary vasoreactivity on testing during right heart catheterization. However, targeted therapies have revolutionized the management of patients for many causes. Targeted therapies modify disease by reversal of the vascular remodeling process, inhibiting platelet aggregation, and causing vasodilatation. The first of these, intravenous epoprostenol, has proven benefits in exercise tolerance, hemodynamics, and survival. Subsequent modifications to improve the stability of the compound have allowed improvement in ease of administration; however, it still remains a continuous infusion with all of the associated risks. Other prostanoids, such as treprostinil (intravenous, subcutaneous, inhaled, and oral formulations) and inhaled iloprost, have subsequently been developed and also appear to be effective in improving functional capacity and survival. Other classes of vasodilators, such as dual endothelin receptor antagonists, selective endothelin receptor antagonists, phosphodiesterase inhibitors, and cyclic guanosine monophosphate antagonists have also been shown to be efficacious in improving functional capacity. These are generally administered orally and are therefore easier to take with fewer logistical issues than continuous infusions. As such, targeted therapy is now more accessible to a wider proportion of patients with pulmonary hypertension, reinforcing the need to make an early diagnosis. Also, combination therapy has been shown to be synergistic and is often used.
Apart from lung transplantation, long-term oxygen therapy is the only measure that has been shown to significantly improve survival in patients with PAH associated with parenchymal lung disease (group 3 PAH). In the diagnostic workup of patients being considered for lung transplantation, particularly in the setting of emphysema, CT permits detection of small primary lung cancers that are not radiographically apparent and can thus significantly influence management. When unilateral lung transplantation is being considered, perfusion scintigraphy is widely used to select the side with the most compromised function.
The treatment of CTEPH is discussed in detail in Chapter 51 .
Manifestations of the Disease
Given the significant crossover in imaging findings of PAH among different subtypes, the remainder of the chapter is organized by specific imaging modalities. As many of the comorbid or inciting disease states, such as acute and chronic pulmonary thromboembolism, intrinsic lung pathologies, and so forth, are discussed separately; specific diagnostic or prognostic findings will be emphasized here.
Radiography
Chest radiography is usually the initial imaging study in patients with suspected pulmonary hypertension. It is useful to assess heart size, pattern of cardiac chamber dilatation, and enlargement of proximal pulmonary arteries, as well as to detect any underlying pulmonary parenchymal disorder.
The radiographic features of pulmonary hypertension consist of central pulmonary arterial dilatation down to the segmental level, with attenuation of peripheral pulmonary blood vessels, giving rise to “peripheral pruning” ( Fig. 53.4 ). Hilar artery enlargement is assessed by measuring the diameter of the interlobar arteries. The upper limit for the transverse diameter of the right interlobar artery measured from its lateral aspect to the bronchus intermedius is 15 mm in women and 16 mm in men. The transverse diameter of the left interlobar artery is difficult to appreciate on the posteroanterior view. On a lateral view the upper limit measured from the circular lucency of the left upper lobe bronchus to the posterior margin of the vessel is 18 mm.

Calcification within the pulmonary arteries may be seen in pulmonary hypertension associated with intracardiac shunts and usually results from prolonged and severe pulmonary hypertension that has resulted in calcific atherosclerosis. Pulmonary artery calcification is usually associated with high PVR and irreversible vascular disease.
Heart size as assessed by cardiothoracic ratio may be normal or enlarged in pulmonary hypertension. Enlargement of the right ventricle causes reduction of the retrosternal clear space on the lateral radiograph. Right atrial dilatation is seen as a widening of the right border of the heart on the frontal projection. Enlargement of the right atrial appendage causes an increase in the retrosternal opacity above the expected location of the right ventricle on the lateral projection.
There is no correlation between the extent of the radiographic abnormalities and the degree of pulmonary hypertension. The accuracy of the chest radiograph in detecting pulmonary hypertension is unknown.
The chest radiograph is useful in defining coexistent conditions, such as pulmonary venous congestion, chest wall and vertebral deformities, emphysema, interstitial lung fibrosis, and other parenchymal and pleural abnormalities. In 90% of patients with idiopathic PAH, the chest radiograph is abnormal at the time of diagnosis. The typical features include enlarged proximal pulmonary arteries with peripheral pruning, cardiomegaly with right-sided chamber enlargement, and pulmonary oligemia ( Fig. 53.5 ). The lungs are otherwise normal, enabling differentiation from pulmonary hypertension secondary to parenchymal lung disease.

Congenital Systemic-Pulmonary Shunts (Congenital Heart Disease)
The radiographic features of pulmonary hypertension caused by chronic shunt physiology include the familiar complex of a dilated pulmonary trunk and central pulmonary arteries. Increased flow results initially in prominence of peripheral pulmonary vessels in proportion to the proximal vessels. On radiographic examination, pulmonary plethora is characterized by vessels being identifiable within 2 cm of the pleural surface. Vasculopathy and the development of pulmonary hypertension may be difficult to identify on the chest radiograph. Peripheral oligemia with rapid tapering of the peripheral vessels and disparity between the proximal and distal calibers is a late manifestation often indicating shunt reversal and Eisenmenger syndrome.
The right ventricle and atrium typically enlarge in proportion to the degree of volume overload. Paradoxically, increased PVR and the development of pulmonary hypertension may lead to reduction in size of the cardiac silhouette.
Atrioseptal defects are characterized by cardiomegaly with enlargement of the right atrium and ventricle ( Fig. 53.6 ); ventriculoseptal defects, by right ventricular hypertrophy and dilatation of the atria and eventually the left ventricle; and PDA, by enlargement of the left atrium, left ventricle, and aortic arch. In addition, there may be calcification of the ductus diverticulum.

Pulmonary Venoocclusive Disease and Pulmonary Capillary Hemangiomatosis
The specific diagnosis of PVOD is suggested radiographically when features of PAH are accompanied by evidence of diffuse pulmonary interstitial edema and a normal-sized left atrium. Back-pressure from the obstructed venular side of the pulmonary circulation results in dilatation of the right-sided heart chambers and central pulmonary arteries, pleural effusions, and signs of pulmonary interstitial edema. The central pulmonary veins and the left atrium are not enlarged, in contrast to patients with mitral stenosis, cor triatriatum, or left atrial myxoma. Mediastinal lymphadenopathy caused by vascular congestion may be present.
In PCH the chest radiograph shows the typical features of pulmonary hypertension. In addition, a reticulonodular or fine nodular pattern may be present, which can suggest an interstitial process.
Left-Sided Heart Disease
Left-sided cardiac diseases, such as myocardial dysfunction, mitral valve disease ( Fig. 53.7 ), and left atrial myxoma, are characterized by pulmonary venous hypertension, interstitial edema, and pleural effusions. Normally, in the erect position the hydrostatic pressure gradient causes higher flow and therefore greater vessel caliber in the dependent lung regions. Pulmonary venous hypertension results in loss or reversal of this normal-caliber gradient, with narrowing of lower lobe and distention of upper lobe vessels. This so-called cephalization, upper zone vascular distention or recruitment, is present in 95% of patients with critical mitral stenosis and 33% to 76% of patients with mild left ventricular dysfunction admitted to coronary care. Upper zone vascular distention is the earliest radiographic feature of pulmonary venous hypertension.

The development of pulmonary interstitial edema is characterized by thickening of the interlobular septa (Kerley B lines) and loss of the sharp margins of vessels and bronchi (perihilar haze and peribronchial cuffing). Finally, frank pulmonary edema results in airspace filling.
The presence of left atrial enlargement can help distinguish left-sided cardiac lesions from PVOD. Hemosiderosis caused by long-standing pulmonary venous hypertension produces fine reticular opacities on chest radiographs. Small (1–3 mm) calcified ossific nodules are the hallmark of mitral stenosis but are now rarely seen. In rare cases a left atrial myxoma is manifested as a heavily calcified mass.
Chronic Thromboembolic Pulmonary Hypertension
Chest radiography in CTEPH reveals the familiar complex of cardiomegaly and right-sided chamber dilatation. The central pulmonary arteries may be asymmetrically enlarged with abrupt cutoff. The peripheral pulmonary vessels are disorganized with regional areas of hypoperfusion and hyperperfusion. Pleural thickening or effusions and peripheral scarring from previous infarction and atelectasis may be present. Please see Chapter 51 for a more complete discussion of CTEPH.
Computed Tomography
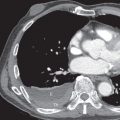
CT is an important tool for the assessment of patients with suspected pulmonary hypertension. The combination of CT pulmonary angiography and high-resolution CT allows a comprehensive assessment of the pulmonary vasculature and lung parenchyma, an assessment of the pattern of cardiac chamber dilatation and the severity of tricuspid regurgitation, and a limited evaluation of right-sided heart function.
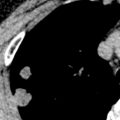
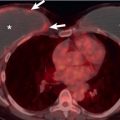
Dilation of central pulmonary arteries is a cardinal sign of pulmonary hypertension on CT and magnetic resonance imaging (MRI) ( Fig. 53.8 ). On CT the diameter of the main pulmonary artery is most frequently measured at the level of its bifurcation at a right angle to its long axis and just lateral to the ascending aorta. The most frequently cited study evaluating absolute pulmonary artery size suggests that a main pulmonary artery of 29 mm or larger on CT is suggestive of pulmonary hypertension with a sensitivity of 69% to 87% and a specificity of 89% to 100%. However, more recent studies have called use of absolute pulmonary artery size into question. A study from 2013 suggested that using a cutoff of greater than or equal to 29.5 mm resulted in only a specificity of 79.4%, and the authors suggested using a cutoff of greater than or equal to 32 mm, which results in a specificity of 90.2%. A study from 2016 demonstrated an even lower specificity of 62% when using the same cutoff, and the authors suggested that such evaluation of absolute pulmonary artery diameter may not be adequate for clinical use. It is our current clinical practice to suggest the possibility of PAH when the main pulmonary artery diameter is greater than or equal to 32.5 mm, while recognizing that a significant number of these patients will be considered falsely positive after evaluation by echocardiography. Of interest, despite these issues with pulmonary artery size, there is some evidence that changes in size over time in an individual patient may have significant prognostic value, with one study demonstrating increasing mortality (hazard ratio, 1.33) for every 1-mm increase in pulmonary artery size over time.