Pulmonary Lymphoid Hyperplasia (Follicular Bronchiolitis)
Etiology, Prevalence, and Epidemiology
Pulmonary lymphoid hyperplasia, also known as follicular bronchiolitis or hyperplasia of bronchus-associated lymphoid tissue, is a benign condition characterized histologically by the presence of polyclonal lymphoid aggregates along the bifurcation of the bronchioles and along the pulmonary lymphatics and is mainly differentiated from lymphocytic interstitial pneumonia (LIP) by its typical distribution limited mainly to the airways. It is seen most commonly in association with connective tissue disease, particularly rheumatoid arthritis; immunodeficiency, particularly acquired immune deficiency syndrome (AIDS) (usually in children); hypersensitivity disorders; and as a nonspecific response in patients with airway infection, airway obstruction, or bronchiectasis.
Clinical Presentation
Pulmonary lymphoid hyperplasia may occur in children and adults (age range, 1.5–77 years). The most common presenting symptoms are progressive shortness of breath and cough. Other manifestations include fever, recurrent pneumonia, and weight loss.
Pathology
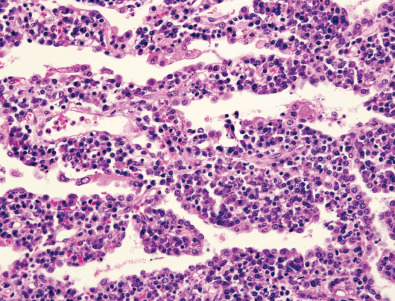
Pulmonary lymphoid hyperplasia is characterized histologically by the presence of discrete foci of hyperplastic lymphoid tissue, often associated with germinal centers, along the bronchovascular bundles ( Figs. 23.1 and 23.2 ). Hyperplastic lymphoid tissue also may be present within interlobular septa and visceral pleura.


Manifestations of the Disease
Radiography
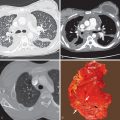
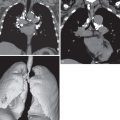
The chest radiograph may be normal or show bilateral reticular or reticulonodular opacities ( Fig. 23.3 ).

Computed Tomography
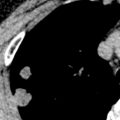
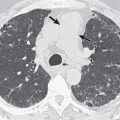
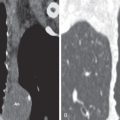
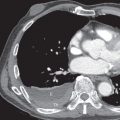
The main high-resolution computed tomography (CT) findings consist of bilateral centrilobular and peribronchial nodules. Most nodules measure less than 3 mm in diameter. Nodules 3 to 12 mm in diameter are present in approximately 50% of patients, and patchy ground-glass opacities are present in approximately 75% of patients; small foci of consolidation may be seen in some patients (see Fig. 23.3 ). The nodules may be diffuse or involve mainly the lower lung zones. The nodules reflect the predominant histologic findings of peribronchiolar inflammation and coalescent germinal centers. Less common manifestations include mild interlobular septal thickening and bronchial dilation.
Differential Diagnosis
Centrilobular nodules are seen in a variety of diseases that affect predominantly the bronchioles or peribronchiolar interstitium. Common causes include hypersensitivity pneumonitis, respiratory bronchiolitis, and infectious bronchiolitis. Pulmonary lymphoid hyperplasia requires lung biopsy for definitive diagnosis.
Synopsis of Treatment Options
Patients with pulmonary lymphoid hyperplasia usually have a good prognosis. Treatment may be directed at the underlying disease or may consist of steroids or azathioprine.
- •
Pulmonary lymphoid hyperplasia or follicular bronchiolitis is a benign condition characterized by the presence of polyclonal lymphoid aggregates mainly along the bifurcation of the bronchioles.
- •
Symptomatic pulmonary lymphoid hyperplasia occurs most commonly in patients with rheumatoid arthritis and immunodeficiency.
- •
The chest radiograph may be normal or show bilateral reticular or reticulonodular opacities.
- •
The main high-resolution CT finding consists of small bilateral centrilobular and peribronchial nodules.
- •
Definitive diagnosis requires surgical lung biopsy. Prognosis is usually favorable.
Lymphoid Interstitial Pneumonia (Lymphocytic Interstitial Pneumonia)
Etiology, Prevalence, and Epidemiology
LIP is a rare condition limited to the lungs and characterized at histology by diffuse infiltration of the alveolar septa by polyclonal lymphocytes. LIP has been considered part of a spectrum of pulmonary lymphoproliferative disorders that range in severity from benign airway-centered cellular aggregates (lymphoid hyperplasia) to malignant lymphomas ; however, immunohistochemical and molecular analyses indicate that the risk of transformation to pulmonary lymphoma is low. Most cases of LIP occur in patients with underlying autoimmune disease or immunodeficiency, most commonly Sjögren syndrome, autoimmune disorders, dysproteinemia, and AIDS; the last-mentioned is particularly seen in children ( Box 23.1 ). When underlying systemic diseases are excluded, a diagnosis of “idiopathic” LIP can be made; however, idiopathic LIP is extremely rare. The clinical and radiologic manifestations of LIP often resemble those of other interstitial pneumonias, and the histologic pattern is that of an interstitial pneumonia that can mimic nonspecific interstitial pneumonia and occasionally hypersensitivity pneumonitis. LIP is part of the updated American Thoracic Society/European Respiratory Society international multidisciplinary consensus classification of idiopathic interstitial pneumonias and is one of the “rare” idiopathic interstitial pneumonias.
Collagen Vascular Disease
Sjögren syndrome
Systemic lupus erythematosus
Rheumatoid arthritis
Polymyositis
Autoimmune Diseases
Hashimoto thyroiditis
Myasthenia gravis
Hemolytic anemia
Pernicious anemia
Autoerythrocyte sensitization syndrome
Chronic active hepatitis
Celiac sprue
Primary biliary cirrhosis
Systemic Immunodeficiency States
HIV/AIDS
Common variable immunodeficiency
Agammaglobulinemia
Drug Induced
Diphenylhydantoin
Captopril
Infections (Other Than HIV)
Legionella pneumonia
Tuberculosis
Mycoplasma
Chlamydia
Epstein-Barr virus infection
Human herpesvirus 8
Miscellaneous
Complication of allogeneic bone marrow transplantation
Pulmonary alveolar microlithiasis
Pulmonary alveolar proteinosis
Idiopathic
AIDS, Acquired immune deficiency disease syndrome; HIV, human immunodeficiency virus.
Clinical Presentation
The average age of presentation is 50 to 60 years; women are affected twice as commonly as men. The onset is typically insidious over several years. The main clinical symptoms are cough, dyspnea, and fatigue. Approximately 80% of patients have serum dysproteinemia, most commonly polyclonal hypergammaglobulinemia. Analysis of bronchoalveolar lavage fluid frequently shows lymphocytosis (30%).
Pathology
LIP is characterized histologically by extensive infiltration of the alveolar septa with polyclonal lymphocytes and variable numbers of plasma cells ( Fig. 23.4 ). Although usually more or less diffuse within the lobule, small nodular foci may be seen as a result of localized proliferation or the formation of germinal centers. Fibrosis is usually absent or mild in severity; occasionally, it may be associated with remodeling and a honeycomb appearance. Airspaces are typically unaffected.