, Joon Woo Lee1 and Jong Won Kwon2
(1)
Department of Radiology, Seoul National University Bundang Hospital, Seongnam, Kyonggi-do, Republic of Korea
(2)
Department of Radiology, Samsung Medical Center, Seoul, Republic of Korea
17.2 Hirayama Disease
17.4 Radiation Myelitis
17.6 Conjoined Nerve Root
17.9.2 Hirayama Disease
17.9.4 Radiation Myelitis
17.9.6 Conjoined Nerve Root
17.9.7 Guillain–Barré Syndrome
Abstract
This chapter will discuss rare spinal disorders which have characteristic imaging findings and mainly involve spinal cord or nerve roots. These include idiopathic spinal cord herniation, Hirayama disease, subacute combined degeneration, radiation myelitis, spinal visceral larva migrans of Toxocara canis, conjoined nerve root, Guillain–Barré syndrome, and hereditary motor and sensory neuropathies.
This chapter will discuss rare spinal disorders which have characteristic imaging findings and mainly involve spinal cord or nerve roots. These include idiopathic spinal cord herniation, Hirayama disease, subacute combined degeneration, radiation myelitis, spinal visceral larva migrans of Toxocara canis, conjoined nerve root, Guillain–Barré syndrome, and hereditary motor and sensory neuropathies.
17.1 Idiopathic Spinal Cord Herniation
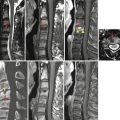
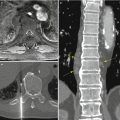
Idiopathic spinal cord herniation is a disorder in which the spinal cord herniates through an anterior dural defect in the upper or middle thoracic level (Parmar et al. 2008). Idiopathic spinal cord herniation is an uncommon clinical entity resulting in progressive myelopathy. On sagittal MR images, an anterior kink of the thoracic spinal cord is seen with focal enlargement of the dorsal subarachnoid space, most commonly between the levels of the T4 and T7 vertebrae. On axial images, part of the spinal cord is seen outside the anterior dura, and the spinal cord appears elongated and rotated at the level of herniation. Sometimes, cerebrospinal fluid can leak into the anterior epidural space, manifest as a thin sliver of fluid signal in the anterior epidural space. In long-standing spinal cord herniation, there can also be bony erosion in the posterior vertebral body due to the herniated spinal cord. On CT myelography, similar findings (anterior kinking of spinal cord on sagittal images, anterior herniation of the spinal cord on axial images) are also evident. Common differential diagnoses include intradural arachnoid cyst, spinal cord adhesion, and arachnoid web with spinal cord deformity. The key to diagnosing spinal cord herniation is to detect the anteriorly herniated spinal cord outside the dura margins on axial images.
17.2 Hirayama Disease
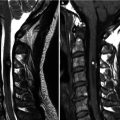
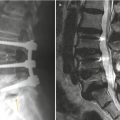
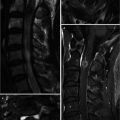
Hirayama disease (juvenile muscular atrophy of distal upper extremity) is a rare form of cervical myelopathy that affects predominantly males in their second and early third decades. Clinical features include muscular weakness and atrophy of the hand and forearm. Interestingly, the brachioradialis muscle is spared; thus the pattern of forearm involvement is referred to as an oblique amyotrophy. It is characterized by progressive muscular weakness and atrophy of the distal upper limbs, followed by spontaneous arrest within several years. Dynamic cord compression during neck flexion with forward displacement of the posterior dura is a characteristic finding. A possible etiology involves disproportional lengths between the vertebral column and the dural canal that may lead to a “tight” dural canal. The normal cervical dura is slack and consists of transverse folds during neck extension. With flexion, the length of the cervical canal increases. In normal patients, the dural slack compensates for the increased length of the cervical canal during flexion. Patients with Hirayama disease may have an imbalance in the growth of the vertebrae and dura resulting in a relatively short dural canal with less ability to compensate; thus the dural canal becomes tight with neck flexion. This results in anterior shift of the posterior dural wall causing spinal cord compression. The pathogenesis of cervical myelopathy may be due to ischemic changes or chronic trauma from repeated neck flexion. Chronic compression may cause microcirculatory disturbances in the anterior portion of the cord, leading to necrosis of the anterior horn cells. Changes are often worst at the C6 vertebral level primarily affecting the anterior horn cells, with cord atrophy developing in later stages of the disease. On MR, flexion sagittal images demonstrate forward displacement of the posterior dura. The posterior epidural space enlarges with flexion and is seen as a crescent of high T1 and T2 signal due to congestion of the posterior internal vertebral venous plexus. On contrast-enhanced images, uniform enhancement of the epidural space with flow voids is seen. On axial images, asymmetric flattening of the spinal cord with forward shifting of the posterior dura can be observed. One article has described loss of attachment of more than two-thirds between the posterior dura and adjacent lamina at the lower cervical level as a reliable imaging finding suggestive of Hirayama disease on MRI performed in the neural position (Chen et al. 2004).
17.3 Subacute Combined Degeneration
The clinical manifestations of vitamin B12 deficiency include hematologic abnormalities such as megaloblastic anemia, while neurologic abnormalities caused by its effect on the brain, peripheral and optic nerves, as well as posterior and lateral columns of the spinal cord are known collectively as subacute combined degeneration (SCD). The most frequent clinical manifestation of vitamin B12 deficiency is SCD of the spinal cord, although it is in itself an uncommon cause of myelopathy (Ravina et al. 2000). The clinical findings of SCD include insidious subacute onset of weakness, paresthesia, loss of vibratory sensation, and sensory ataxia. Symptoms are usually symmetric and progress from distally to proximally and may progress to paraplegia in untreated cases. Timely diagnosis and appropriate treatment is important to prevent irreversible neurologic injury. Vitamin B12 cobalamin is a complex molecule found in food from animal and some plant sources. Following ingestion, vitamin B12 is bound to intrinsic factor (IF) secreted by gastric parietal cells; the B12–IF complex then binds to the mucosa of the terminal ileum where B12 is absorbed. Any abnormality along this pathway may lead to vitamin V12 deficiency. However, because of substantial total body stores of 2–5 mg, the effects of vitamin B12 deficiency from malabsorption syndromes such as pernicious anemia, regional enteritis, tropical sprue, celiac disease; surgical procedures such as gastrectomy and ileal resection; as well as inadequate intake in strict vegetarians may not develop for several years.
The cause of vitamin B12-associated neurologic disease is consequent to increased myelinolytic tumor necrosis factor-α (TNF-α) and decreased neurotrophic factor levels. Vitamin B12 deficiency leads to an overproduction of TNF-α and downregulation of two neurotrophic factors: epidermal growth factor (EGF) and interleukin-6 (Scalabrino et al. 2004). Vitamin B12-associated myelopathy is histologically characterized by multifocal demyelination, vacuolization, and axonal degeneration typically involving the dorsal and lateral columns and occasionally the ventral column (Timms et al. 1993). Linear T2 hyperintensities appearing as an “inverted V” in the posterior column of the spinal cord is a characteristic finding on MR (Timms et al. 1993). In some patients, bilateral paired nodular T2 hyperintensities described as “dumbbell”- or “binocular”-shaped areas are seen.
17.4 Radiation Myelitis
Radiation can be a cause of myelopathy (Calabro and Jinkins 2000). On MR images, radiation myelitis shows similar features to other non-tumorous myelopathy such as acute transverse myelitis or multiple sclerosis. These features include patchy T2 hyperintensities and patchy enhancement within the spinal cord. The clue that points to radiation myelitis as a cause is a history of previous radiation therapy to the involved segment. Fatty marrow signal change in the vertebral bodies at the same region is also sometimes observed. In the acute phase of radiation myelitis, the spinal cord is enlarged and demonstrates T1 hypointensity and T2 hyperintensity. Contrast enhancement is variable and may be patchy or ringlike. In the chronic phase, the affected portions can become atrophic.
17.5 Spinal Visceral Larva Migrans of Toxocara canis
Humans are manifested by Toxocara canis as visceral larva migrans when uncooked cow or cattle liver or meat containing the infective form of Toxocara canis is ingested. Spinal visceral larva migrans of Toxocara canis is an unusual cause of non-tumorous myelopathy. Myelopathy is secondary to hematogenous infestation of the spinal cord with visceral larva migrans. There may be migration from pulmonary lesions. Typical MR findings of spinal visceral larva migrans are single lesion, short segmental involvement of T2 hyperintensity with mild cord swelling, and focal nodular enhancement on posterior or posterolateral columns after gadolinium administration (Lee et al. 2010). The diagnosis of toxocariasis relies on high titers of Toxocara canis antibodies, eosinophilia in blood and/or cerebrospinal fluid, and demonstration of an intrathecal synthesis of anti-Toxocara canis antibodies. The clinical and radiologic improvement as well as the normalization of the cerebrospinal fluid parameters after anti-helminthic therapy supports the diagnosis (Xinou et al. 2003).
17.6 Conjoined Nerve Root
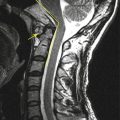
A conjoined nerve root is composed of two adjacent nerve roots which share a common dural envelope at some point along their course from the dural sac. The prevalence of lumbosacral nerve root anomalies varies widely. Myelography reports of patients complaining of leg pain and suspected disk herniation reveal an incidence of suspected nerve root anomalies in 4 % (Kadish and Simmons 1984). Anatomic studies have demonstrated a much higher incidence (Kikuchi et al. 1984).
Conjoined nerve roots are commonly found between L5–S1 and S1–2 (Song et al. 2008). Conjoined nerve root may be divided into various types. The most clinically relevant pattern is one where the common nerve root outside the dura bifurcates into an upper nerve coursing in a horizontal direction occupying the lower part of the neural foramen and a lower nerve coursing in a more vertical direction just posterior to the upper nerve in the epidural space. In such a pattern, the upper nerve can be easily compressed by a mild disk bulge and mild facet arthropathy due to its horizontal course. The lower nerve can be also compressed by the facet joint along its vertical course. If the presence of a conjoined nerve root is not discovered before operation, the nerve root may be injured during retraction of the dural sac or could even be mistaken for a herniated disk and resected. We suggest three radiologic signs on axial MR images at the disk level useful for diagnosing conjoined nerve roots preoperatively: these are as follows: (1) corner sign, asymmetric morphology of the anterolateral corner of the dural sac; (2) fat crescent sign, intervening extradural fat tissue between the asymmetric dural sac and conjoined nerve root; and (3) parallel sign, visualization of the parallel course of the entire nerve root at the disk level (Song et al. 2008).
17.7 Guillain–Barré Syndrome
Guillain–Barré syndrome is a relatively common acute, and rapidly progressive, inflammatory demyelinating polyneuropathy. On contrast-enhanced MR, there is marked enhancement of the thickened nerve roots at the conus medullaris and of the cauda equina. The enhancement can be confined to the anterior roots or involve the anterior and posterior nerve roots (Byun et al. 1998; Berciano 1999; Georgy et al. 1994; Iwata and Utsumi 1997). The spinal cord and nerve roots in the thecal sac normally do not enhance because of the blood–nerve and blood–brain barriers. Other causes of intradural nerve root enhancement are leptomeningeal metastases, lymphoma, chronic inflammatory demyelinating polyneuropathy (CIDP), sarcoidosis, AIDS-related cytomegalovirus polyradiculopathy, Lyme disease, and postoperative arachnoiditis. Diffuse enhancement of intradural nerve roots with a history of acute progressive ascending and symmetric paralysis of the extremities strongly suggest Guillain–Barré syndrome.
17.8 Hereditary Motor and Sensory Neuropathies (HMSN)
Hereditary motor and sensory neuropathies (HMSN) are a heterogeneous group of genetically determined peripheral neuropathies characterized by symmetrical and predominately distal motor and sensory disturbances and a slowly progressive course.
HMSN is classified into three types: HMSN type 1 (Charcot–Marie–Tooth, CMT type 1), HMSN type 2 (CMT type 2), HMSN type 3 (Dejerine–Sottas disease, DSD). CMT type I and DSD are the disorders most characteristically associated with marked thickening of peripheral nerves (hypertrophic neuropathies).
Charcot–Marie–Tooth (CMT) disease is the most common inherited neuromuscular disorder. CMT disease can be further divided into two types based on the electrophysiological and pathological findings. CMT disease type 1 is the demyelinating form and is characterized by a slow motor median nerve conduction velocity. CMT disease type 2 is the axonal form and shows a normal or slightly reduced nerve conduction velocity (Gaeta et al. 2012).
On spine MR, diffuse thickening of the intradural nerve roots and extradural spinal nerves is a characteristic finding suggestive of HMSN. On postcontrast images, the intradural nerve roots can be enhanced in HMSN (Cellerini et al. 2000). Diffuse hypertrophy of the cauda equine and spinal nerves are also seen in chronic inflammatory demyelinating polyneuropathy (CIDP) (Vallat et al. 2010). The differentiation between HMSN and CIDP is difficult by MRI findings only.
Forget Me Nots!
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


