CHAPTER 63 Restrictive Cardiomyopathy
Restrictive cardiomyopathy (RCM) is the least common cardiomyopathy, and is characterized by diastolic dysfunction with restrictive ventricular filling with normal or near-normal systolic function and wall thickness.1 RCM may be idiopathic or associated with other infiltrative diseases, such as amyloidosis, endomyocardial disease, sarcoidosis, iron deposition disease, and storage diseases. Numerous other diseases may have a prominent restrictive component. Presentation of RCM is variable, and diagnosis is often difficult. The prognosis for most forms of RCM is poor, and it is important to distinguish RCM from constrictive pericarditis, which may have a similar clinical presentation
CARDIAC AMYLOIDOSIS
Prevalence and Epidemiology
Primary amyloidosis is a rare but devastating disease, with an incidence of 9 per 1 million and mean survival of approximately 13 months after diagnosis.2 Cardiac involvement in primary amyloidosis is common, with 60% of patients exhibiting ECG or echocardiographic abnormalities. Death is attributed to cardiac causes in at least 50% of patients with primary amyloidosis who die either from heart failure or from a malignant arrhythmia.2
Senile amyloidosis predominantly affects men older than 70 years and involves the heart in 25% of individuals older than 80 years.3 Senile cardiac amyloidosis is often clinically silent; however, extensive amyloid deposition can lead to significant clinical symptoms.
Etiology and Pathophysiology
Amyloid deposits, regardless of their protein composition, all have a characteristic appearance on light microscopy, staining pink with Congo red dye and exhibiting apple-green birefringence under polarized light microscopy. Nearly all organ systems can be involved, including the kidneys, heart, blood vessels, central and peripheral nervous system, liver, bowel, lungs, eyes, skin, and bones. Cardiac amyloidosis is a devastating progressive process that leads to congestive heart failure, angina, and arrhythmias.1
In addition to mechanical effects on myocardial stiffness, amyloid deposition induces oxidative stress that depresses myocyte contractile function. Myocardial ischemia may also result from microvascular disease. Amyloid deposits typically spare the epicardial vessels, whereas involvement of intramyocardial vasculature is seen in more than 90% of patients with AL amyloidosis.4
Manifestations of Disease
Imaging Techniques and Findings
Ultrasonography
Increased wall thickness without dilation of the ventricular cavity and preserved systolic function until relatively advanced stages of the disease are hallmarks of cardiac amyloidosis on echocardiography (Fig. 63-1). Amyloid deposits may involve nearly all regions of the heart, including valves, myocardium, interatrial septum, and pericardium, and manifestations of this involvement can be seen as multivalvular regurgitation, thickening of the interatrial septum, atrial dilation, pericardial effusions, and diffuse thickening of the right ventricular and left ventricular (LV) myocardium.5 A classic finding of myocardial amyloidosis is a granular sparkling pattern on two-dimensional echocardiography. This pattern is not specific for amyloidosis, however, and can be seen in patients with hypertensive cardiomyopathy, glycogen storage disorders, and hypertrophic cardiomyopathy. Atrial and ventricular thrombi are common findings, particularly in advanced disease.
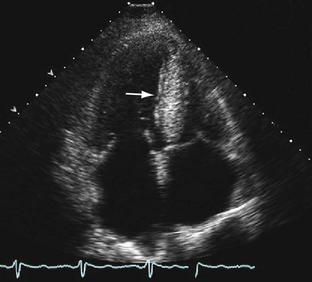
 FIGURE 63-1
FIGURE 63-1Pulsed wave Doppler echocardiography is helpful in assessing diastolic dysfunction in cardiac amyloidosis. The initial diastolic abnormality is abnormal relaxation (grade 1 diastolic dysfunction) resulting from increased ventricular wall thickness; the pattern becomes restrictive (grades 3 to 4) when progressive amyloid infiltration decreases LV compliance and increases left atrial pressure. The filling pattern may normalize temporarily (pseudonormalization—grade 2 diastolic dysfunction) as a result of combined relaxation abnormality and moderate increase in left atrial filling pressure before becoming frankly restrictive. Deceleration time is an important prognostic variable in cardiac amyloidosis; the average survival for patients with a deceleration time less than 150 ms is less than 1 year versus 3 years for patients with a deceleration time greater than 150 ms.6 A combination of LV wall thickness greater than 15 mm and fractional shortening of less than 20% (thought to reflect combined systolic and diastolic dysfunction) is associated with a median survival of 4 months.7 Right ventricular function has also been correlated with poor prognosis in patients with cardiac amyloidosis.8
Tissue Doppler imaging (TDI) has emerged more recently as a useful technique in assessment of LV regional wall motion and diastolic dysfunction in patients with cardiac amyloidosis. Koyama and colleagues9 showed that TDI measurements differentiated patients without from patients with cardiac amyloidosis, and amyloidosis patients with and without heart failure. TDI more clearly documented diastolic function than conventional Doppler-derived indices. Myocardial strain and strain rate imaging have also been investigated in cardiac amyloidosis, and these techniques have documented early impairment in systolic function before the onset of clinical heart failure.10
Computed Tomography
ECG gated contrast-enhanced CT reveals many of the findings discussed previously with regard to echocardiography: ventricular thickening with preserved systolic function, enlarged atria, and pericardial and pleural effusions (Fig. 63-2). Little evidence currently available suggests that CT is useful in the initial diagnosis of cardiac amyloidosis or in distinguishing amyloidosis from other causes of RCM.
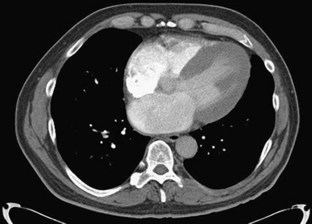
 FIGURE 63-2
FIGURE 63-2Magnetic Resonance Imaging
MRI has shown considerable promise in diagnosis and characterization of cardiac amyloidosis.11–14 Cine steady-state free precession (SSFP) images readily show findings of ventricular thickening with normal chamber size, atrial enlargement, and preserved systolic function (Fig. 63-3). Pleural and pericardial effusions are common and are well depicted on MRI. Impaired diastolic relaxation is often appreciated on cine SSFP images, and mitral inflow measurements can be obtained using cine phase contrast pulse sequences to obtain information analogous to Doppler echocardiography.
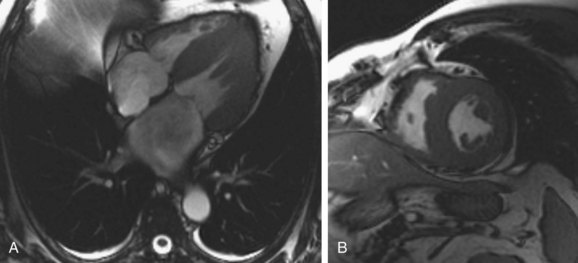
 FIGURE 63-3
FIGURE 63-3After administration of contrast medium, striking abnormalities are often seen on MDE pulse sequences, with patients with cardiac amyloidosis typically showing diffuse irregular hyperenhancement in noncoronary distributions (Fig. 63-4). A circumferential subendocardial hyperenhancement pattern has been described and correlated with predominant amyloid deposition in the subendocardial myocardium; however, in our experience, patterns of hyperenhancement are quite variable. Right ventricular late enhancement is a notable feature of amyloidosis and can help to distinguish this from hypertrophic cardiomyopathy with foci of enhancing fibrotic tissue.
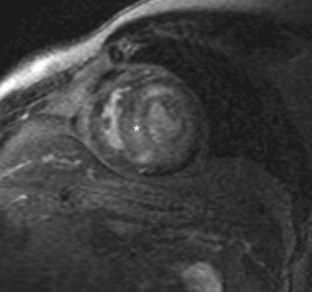
 FIGURE 63-4
FIGURE 63-4Abnormalities of myocardial nulling are also common in amyloidosis and can help to distinguish this disease from other pathologies. A cine multi-TI inversion recovery sequence, in which each image or phase is acquired with a slightly longer inversion time (TI), is often used to select the optimal TI for the delayed enhancement acquisition. As TI increases, blood and myocardium pass through a null point at which signal is minimized. Generally, the blood pool contains a higher concentration of gadolinium, has a shorter T1 relaxation time, and passes through the null point before myocardium. In many amyloid patients, this progression is reversed, with myocardial tissue reaching the null point before the blood pool (Fig. 63-5).
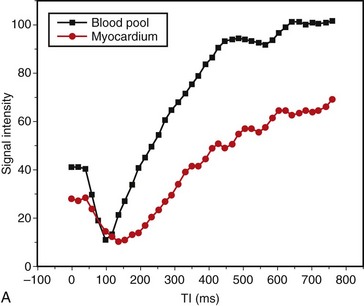
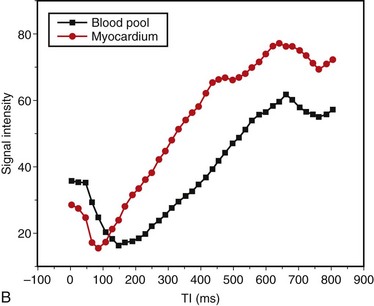
 FIGURE 63-5
FIGURE 63-5Nuclear Medicine
Nuclear medicine has generally played a minor role in the diagnosis and characterization of cardiac amyloidosis. Various tracers, mainly phosphonates, have been used to assess patients with suspected cardiac involvement, with heterogeneous results. More recent developments include the use of Tc 99m DPD scintigraphy to distinguish between AL and senile or transthyretin-related cardiac amyloidosis in patients with known cardiac amyloidosis.15
EOSINOPHILIC ENDOMYOCARDIAL DISEASE
Prevalence and Epidemiology
It is unclear whether these are two distinct diseases or different manifestations of the same underlying pathologic process. IHES occurs in temperate countries, has a more aggressive and rapidly progressive course, and is related to hypereosinophilia, whereas endomyocardial fibrosis occurs most commonly in equatorial Africa and is not definitely related to hypereosinophilia. Endomyocardial fibrosis occurs mainly in children and adolescents belonging to the poorest groups of the population and is an endemic cause of heart disease in certain tropical areas, such as the coastal regions of Mozambique, where 9% of the population is affected.16 The etiology of endomyocardial fibrosis is unclear; however, in most cases eosinophilia is seen in the initial inflammatory process, and histologically the endocardial lesions are similar to those seen in IHES. An alternative hypothesis has focused on an animal protein–deficient cassava diet, which has induced endomyocardial fibrosis in an animal model.



