The overall incidence of a lethal skeletal dysplasia is 1/10,000. A detailed fetal anatomic survey is required to satisfactorily evaluate a fetus for a suspected skeletal dysplasia. However, specific key measurements, ratios, and anatomic landmarks will aid the sonographer/sonologist in determining not only if a skeletal dysplasia is present, but also if it is lethal.
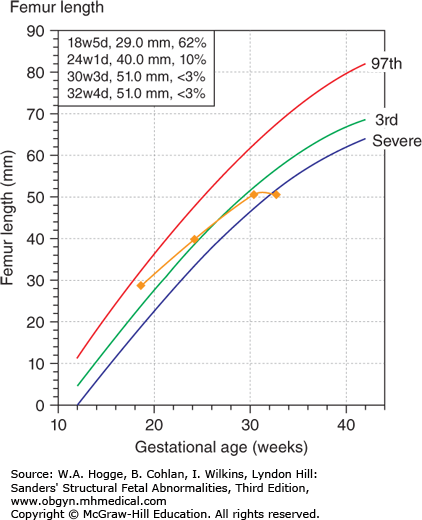
Because of the rarity of lethal skeletal dysplasias, a femur length less than 2 SD (standard deviations) of the mean is not necessarily diagnostic. However, when the femur length is 5 mm below 2 SD, the diagnosis of a skeletal dysplasia can be confirmed.
When a skeletal dysplasia is suspected, all of the long bones should be measured bilaterally to exclude a focal skeletal abnormality.
The appearance of the long bones (i.e., bowing, fractures, or degree of mineralization) may provide important clues in classifying the type of skeletal dysplasia.
Pre- or postaxial polydactyly and clubbed feet are associated with specific skeletal dysplasias.
The femur length/abdominal circumference ratio is normally between 0.20 and 0.24. A ratio of less than 0.16 in a population at risk is consistent with a severe skeletal dysplasia.
In a normal fetus, the femur and foot are of comparable length. A femur length/foot ratio of less than 0.87 discriminates between intrauterine growth restriction and a severe skeletal dysplasia.
A thoracic circumference at the level of the four-chamber view below fifth centile or a thoracic/abdominal circumference ratio less than 0.83 is consistent with severe skeletal dysplasia. A narrow chest and protruding abdomen subjectively suggest a diagnosis of severe skeletal dysplasia.
The presence of severe femur length shortening, in isolation, is insufficient to diagnose a severe skeletal dysplasia. Utilizing the previous criteria, the sonographic detection of lethal skeletal dysplasias is greater than 90%. However, the ability to accurately diagnosis the type of severe skeletal dysplasia is only 50%. A thorough postdelivery evaluation of the fetus is therefore required to confirm a diagnosis and to provide appropriate counseling to the family.
Definition Achondrogenesis is a group of lethal skeletal dysplasias characterized by severe limb and trunk shortening, with a disproportionately large head.
Epidemiology Achondrogenesis is rare.
Embryology There are two main subtypes. In type I, there is gross shortening of the limbs (micromelia) with flaring of the ribs, or fractures, and lack of ossification of the calvarium and spine. Micrognathia may be present. Hydrops may occur. Type I is subdivided into type IA, which has multiple rib fractures, and type IB, in which rib fractures do not occur. In type II (Langer-Saldino), there is no flaring of the ribs or rib fractures, and there is a relatively normal skull ossification, but otherwise the features are similar (i.e., severe micromelia, trunk shortening, edema of the soft tissues, and a disproportionately large head). Ossification of the lumbar vertebrae is minimal (type I) or absent (type II), and the sacral, pubic, and ischial bones show little ossification. Characteristic craniofacial features include micrognathia, flat face, and frontal bossing. Type II is occasionally associated with other abnormalities, including cleft palate, postaxial polydactyly of the feet, cystic hygroma, and cardiac defects. Type IA is caused by mutations in the thyroid hormone receptor interactor gene. Mutations in the diastrophic dysplasia sulfate transporter (DTDST) gene cause achondrogenesis type IB. Both type IA and type IB are inherited in an autosomal recessive fashion. Molecular defects in type II collagen have been found in cases of type II achondrogenesis with dominant inheritance. Consultation with a geneticist or genetic counselor is recommended when this group of disorders is suspected.
Inheritance Patterns Type I is inherited as an autosomal recessive disorder. Type II achondrogenesis is a new dominant mutation. Recurrence in some families has been reported, likely due to germ cell mosaicism.
Teratogens None are known.
Prognosis Achondrogenesis is lethal antenatally or in the newborn period.
Decreased/absent fetal movement
Extremities
Micromelia
Brachydactyly
Clubfeet
Club hands
Long bones poorly calcified
Both sides of the long bones are visualized (no acoustic shadowing).
Because of significant demineralization, some bones may not be identifiable sonographically.
Head
Short neck
Midface hypoplasia
Normal-to-absent calcification of the skull
Cystic hygroma
Chest
Small, bell-shaped ribs that do not extend beyond the cardiac atrium
Rib fractures
Pulmonary hypoplasia
Protuberant abdomen
Spine: normal-to-poor calcification
First-trimester increased nuchal translucency
Microcephaly
Cleft palate
Ventricular septal defect or other cardiac defects
Pyelocaliectasis
Hydrops
Three-dimensional (3-D) imaging accurately portrays facial features, brachydactyly, and the relative disproportion of limb segments.
Rib fractures may help to differentiate type IA.
Investigations and Consultations Required There are two goals of further investigations: determining whether the skeletal dysplasia is lethal and, if possible, determining which skeletal dysplasia the case represents. If the precise skeletal dysplasia is uncertain, other investigations are warranted, including a fetal echocardiogram. Congenital heart disease is a feature of a number of skeletal dysplasias and may be seen in type II achondrogenesis. Once a diagnosis of a lethal condition is established, and if the pregnancy is beyond the legal limits for termination, the family should meet with a neonatologist to discuss neonatal management.
Monitoring Further ultrasound examinations looking for relative growth and mineralization of the spine, ribs, and long bones may help to establish the diagnosis. Once a diagnosis of a lethal condition has been established, routine supportive maternal care should be provided. No attempts to treat preterm labor or to monitor fetal status are appropriate.
Pregnancy Course Polyhydramnios is a common complicating factor and may result in preterm labor. In addition, nonimmune hydrops may develop. If the polyhydramnios is significant and symptomatic, delivery should be undertaken. No tocolysis should be administered for preterm labor.
Pregnancy Termination Issues An intact fetus must be delivered, and the postmortem examination, including radiologic, morphologic, biochemical, and molecular studies, must be performed by individuals with extensive experience in skeletal dysplasias. Determining a precise diagnosis is essential to counseling the family regarding recurrence risk and for offering the appropriate prenatal diagnosis in subsequent pregnancies.
Delivery Vaginal delivery without electronic monitoring is appropriate. Some forms of achondrogenesis have relative macrocephaly, which may make vaginal delivery difficult. Delivery should occur in a center with expertise in the diagnostic evaluation of skeletal dysplasias or one with an interest and expertise in fetal pathology.
Neonatal Resuscitation When the diagnosis is known prior to delivery, a prenatal decision for nonintervention, made after discussion with the family, is appropriate. If the diagnosis is uncertain, it is appropriate to support respiration to allow time for diagnostic evaluation and parental adaptation to occur.
Neonatal Transport Referral to a tertiary perinatal center is appropriate for confirmation of a diagnosis. Mechanical ventilatory support is often required during the transport.
Testing and Confirmation Postnatal radiographs can help determine the specific type of achondrogenesis.
Nursery Management Confirmation of the diagnosis, comfort care for the infant, and counseling and support of the family are the primary goals. Provision of mechanical life support in the interim is appropriate. Live-born infants that do not receive life support usually do not survive beyond the immediate newborn period.
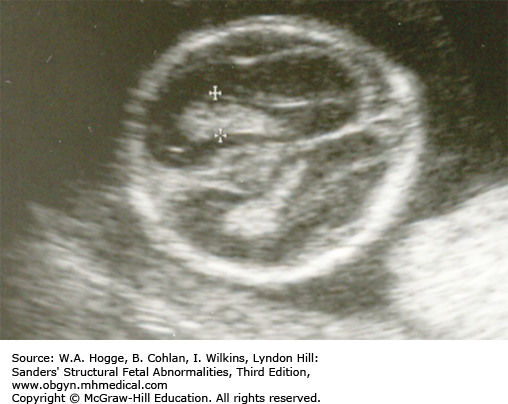
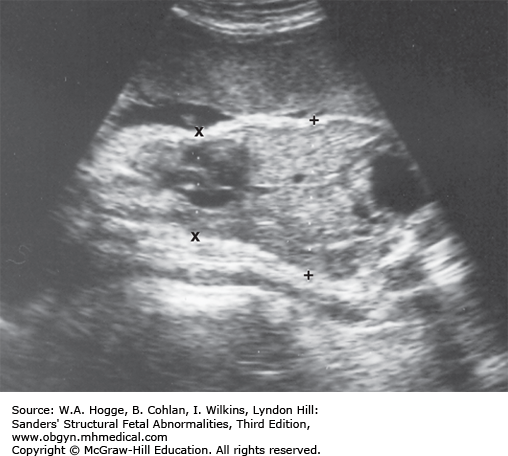
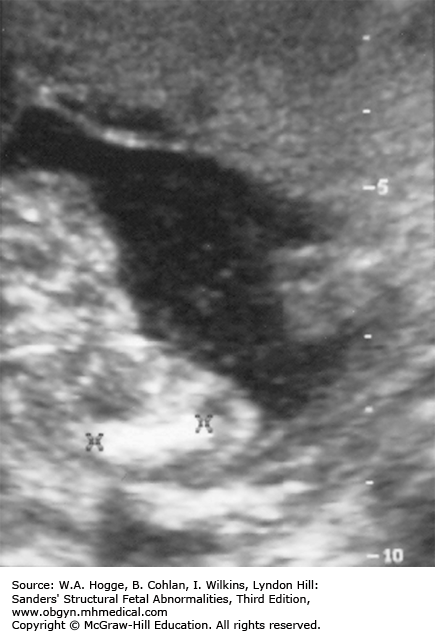
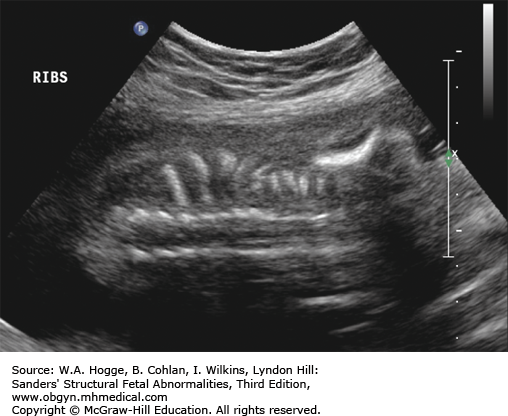
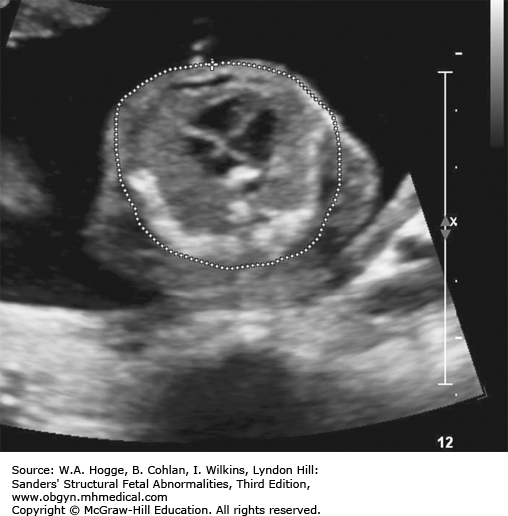
Definition Achondroplasia is the most common skeletal dysplasia and is characterized by rhizomelic limb shortening with macrocephaly.
Epidemiology Occurrence is 1 in 15,000 live births (M1:F1).
Embryology The phenotype is due to decreased endochondral ossification and is characterized by rhizomelic micromelia, macrocephaly with frontal bossing, and midface hypoplasia. The macrocephaly is due to anatomic megalencephaly secondary to an increase in the number of brain cells. Achondroplasia is due to mutations in the fibroblast growth factor receptor 3 (FGFR3) gene that maps to the distal end of chromosome 4p. Almost all cases demonstrate the same single base-pair substitution.
Inheritance Patterns Inheritance is autosomal dominant, with nearly 90% of cases representing new gene mutations. Recurrence risk in subsequent pregnancies is approximately 5% because of germ cell mosaicism.
Teratogens None are known.
Prognosis Intelligence and life span are usually normal. Affected individuals are at risk for neurologic complications, including spinal cord compression at the foramen magnum and the thoracolumbar region. Other complications include obstructive sleep apnea and obesity. The average life expectancy is decreased by 15 years. Homozygosity for achondroplasia, which occurs when both parents are affected, is lethal, with either stillbirth or early neonatal death from pulmonary hypoplasia.
Head
Relative macrocephaly
Frontal bossing
Depressed nasal bridge
Midface hypoplasia
Chest
Small, but within the normal range
Extremities
Mild-to-moderate rhizomelic shortening
Femur length initially within the normal range
Late second and early third trimester: femur length between –2 and –3 SD
Bowing of femur
Hands/feet
Brachydactyly (short fingers)
Trident hand
Separation of second and third digit
Inability to approximate third and fourth fingers
Equal-length fingers
Spine
Lordosis
Central nervous system
Ventriculomegaly
Ultrasound does not provide a firm diagnosis until the third trimester.
When achondroplasia is suspected, a femur length/abdominal circumference (FL/AC) ratio of less than 0.16 in the third trimester is not a reliable indicator of severe skeletal dysplasia.
Severe intrauterine growth restriction: The limbs will not be as short, and there may be markedly decreased fluid.
Thanatophoric dwarfism: Similar appearance, but it develops earlier and is much more severe (see Section 8.14).
The pattern of long-bone growth will readily distinguish these two diagnoses.
With thanatophoric dysplasia, long-bone shortening begins in the first trimester, in contrast to the late second or early third trimester with achondroplasia.
Investigations and Consultations Required Achondroplasia may present with mild shortening of the long bones in fetal life, and there is significant overlap with normal. Unless the fetus is at risk (one of the parents has achondroplasia), this would represent a new mutation, and the diagnosis may be in doubt. As such, an evaluation for other causes of shortened long bones, particularly early-onset intrauterine growth restriction, should be undertaken. An amniocentesis for DNA testing, looking for the most common mutations causing this disorder, may be useful. Fetal echocardiography and chromosomal studies should be performed to exclude other conditions with mild shortening of the long bones. Consultation with a pediatric geneticist should be done to assist the family with an understanding of the disorder and the implications for further care.
Monitoring Serial ultrasound examinations looking for growth of the long bones may be necessary to make the diagnosis, but once it is made, no further exams are needed unless obstetrical complications arise. No change in standard obstetric practice is necessary.
Pregnancy Course Mild polyhydramnios may develop, but preterm labor or other obstetric complications are unusual.
Pregnancy Termination Issues Generally, this diagnosis is not made until the third trimester. It may be made sooner in families at risk, but for those individuals, termination of pregnancy is generally not desired. If the termination is contemplated and a DNA diagnosis to confirm is available, any method of termination is available. If the diagnosis is in doubt, an intact fetus and an autopsy, as well as x-ray studies, would be necessary.
Delivery Delivery should occur in a tertiary center with full capabilities for neonatal resuscitation in the event of an incorrect diagnosis. Because of the narrow foramen magnum and upper cervical spine, spinal cord compression is a significant risk with neck manipulation. Respiratory distress in the neonatal period is not uncommon, also necessitating a tertiary center for delivery.
Resuscitation In the absence of known homozygosity, there are no contraindications to full resuscitative efforts following delivery if there is delay in spontaneous onset of respiration.
Transport The primary indication for referral in the immediate neonatal period is for confirmation of the diagnosis and evaluation by pediatric subspecialists, including genetics, orthopedics, and neurology.
Testing and Confirmation Postnatal skeletal radiographs are pathognomonic and can confirm the clinical phenotype.
Nursery Management Hydrocephalus may develop, although not usually in the neonatal period.
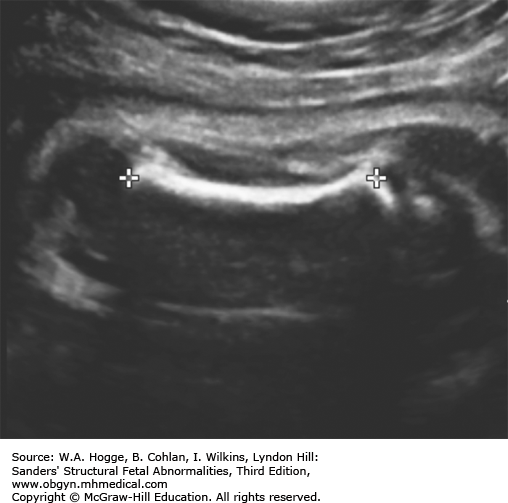
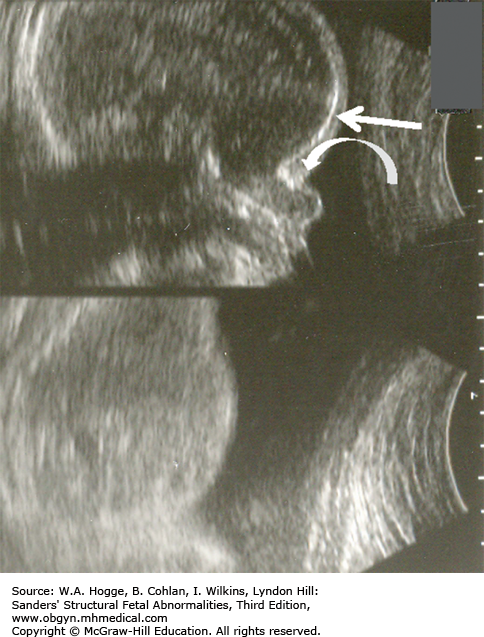
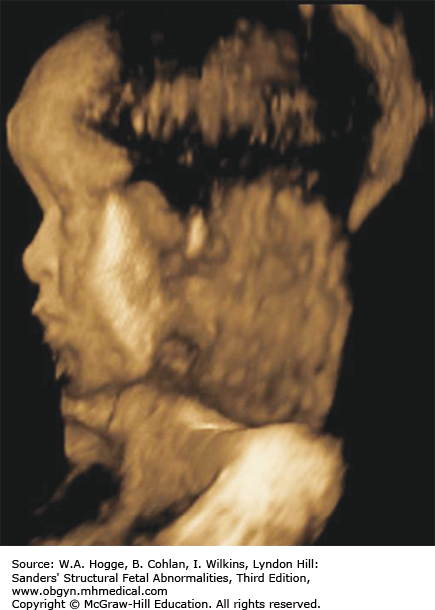

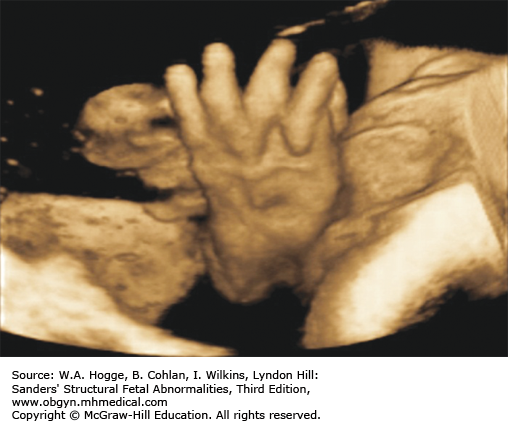
Definition Arthrogryposis is a term for a group of disorders characterized by congenital, usually nonprogressive, joint contractures at multiple sites.
Epidemiology Occurrence is 1 to 3 in 10,000 births (M1:F1).
Embryology Arthrogryposis is a physical finding that results from a heterogeneous group of disorders. The arthrogryposes can be divided into three groups: (1) limb involvement only; (2) generalized neuromuscular involvement; and (3) central nervous system, as well as neuromuscular, involvement. There are more than 300 genetic, chromosomal, and sporadic multiple-malformation syndromes that have congenital contractures as a feature. Possible etiologies include a vascular event, neurologic or myopathic process, connective tissue abnormalities, chromosomal abnormalities, intrauterine compression, or a teratogenic exposure. Only one-half of children with arthrogryposis receive a specific diagnosis. Trisomy 18 is the most common chromosomal diagnosis. Myotonic dystrophy is an autosomal dominant disorder with a severe arthrogrypotic congenital form. It occurs only in offspring of mothers with myotonic dystrophy.
Inheritance Patterns Autosomal dominant, recessive, and X-linked syndromes have been described.
Teratogens Maternal hyperthermia and congenital infections are teratogenic.
Prognosis The prognosis is dependent on an accurate diagnosis and may range from perinatal lethal disorders to those with only mild-to-moderate orthopedic limitations.
Extremities
Limited joint mobility
May involve one or more joints
Clubfeet
Clenched hands
Ulnar deviation
Extension at the elbow
Flexed wrists
Hypoplastic muscles
Head/Neck
Fixed hyperextension
Micrognathia
Umbilical cord
Short, due to decreased movement
Polyhydramnios
Intrauterine growth restriction
First-trimester thickened nuchal translucency
Cleft palate
Ventriculomegaly
Microcephaly
Agenesis of the corpus callosum
Holoprosencephaly
Pulmonary hypoplasia
Pleural effusions
Hydrops
Tracheoesophageal fistula
Scoliosis
Oligohydramnios
The hallmark is bilateral clubbed feet and decreased fetal movement.
Diagnosis usually is made in the second trimester.
Four-dimensional (4-D) ultrasound permits the diagnosis of motor failure, even in the latter part of the first trimester.
In some cases, there are only mild orthopedic limitations.
Pena-Shokeir syndrome: a perinatal lethal form of multiple contractures
There is nuchal thickening or generalized edema in all cases of Pena-Shokeir syndrome.
The stomach is small to absent due to decreased swallowing.
While pulmonary hypoplasia is always present with Pena-Shokeir syndrome, it does not occur in mild forms of arthrogryposis.
Investigations and Consultations Required The extent and direction of further evaluation will depend in part on associated ultrasound findings, which suggest whether the arthrogryposis is isolated or part of a syndrome. Fetal karyotype, microarray analysis, and studies to rule out congenital infections should be done, especially if additional malformations are detected. Fetal echocardiography may also be helpful in establishing a diagnosis. Consultations with a medical geneticist should be done to determine if specific molecular testing should be done.
Monitoring Standard obstetric care is appropriate unless there are features consistent with the lethal forms of the disorder. Serial ultrasound examinations to assess the possible progression of limb abnormalities may be helpful for prognostic purposes. The presence or absence of polyhydramnios and nonimmune hydrops can also be evaluated during these follow-up ultrasound examinations.
Pregnancy Course No changes are expected. The finding of severe polyhydramnios is highly suggestive of the lethal forms of the condition, and amnioreduction, except for maternal indications, should not be considered. If a lethal form of arthrogryposis is suspected, consultation with a neonatologist to discuss neonatal management is appropriate.
Pregnancy Termination Issues Arthrogryposis may be part of a heterogeneous group of disorders; a complete autopsy on an intact fetus should be performed. Accurate recurrence risk counseling requires a precise diagnosis.
Delivery Breech presentation is common in fetuses with arthrogryposis. The mode of delivery should be based on obstetric indications. Delivery at a tertiary center is appropriate as an exact diagnosis may not be established prenatally, and a team prepared to handle complex neonatal issues should be available.
Resuscitation The decision not to offer resuscitation is easiest when there is a definite diagnosis of a concomitant lethal condition. In all other circumstances, resuscitation should be initiated and support continued, if needed, while a diagnostic evaluation is completed.
Transport Referral to a tertiary perinatal center for diagnostic evaluation is appropriate. With an otherwise-asymptomatic infant, referral for outpatient evaluation and treatment planning is also acceptable.
Testing and Confirmation Because the long-term prognosis is heavily influenced by associated or coexisting disorders, complete diagnostic evaluation and etiologic determination are the top priorities. Muscle biopsy and nerve conduction studies are important in determining the potential for habilitation.
Nursery Management Treatment is directed toward achieving stable, functional weight-bearing and manual dexterity. Initially, positioning, physical therapy, and bracing are used, followed by surgery in later infancy and childhood.
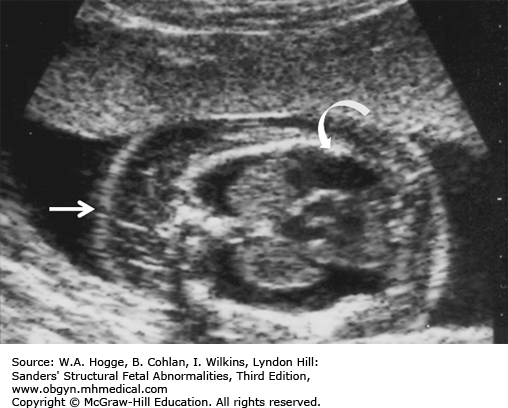
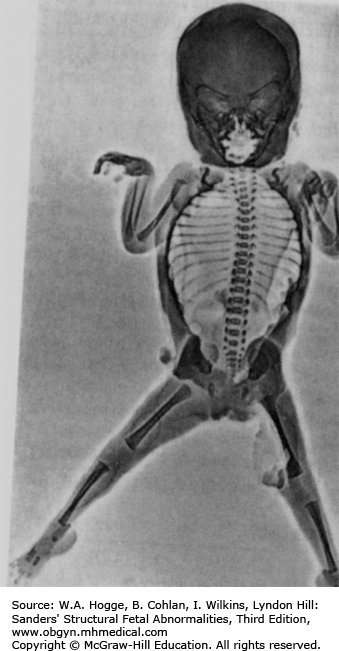
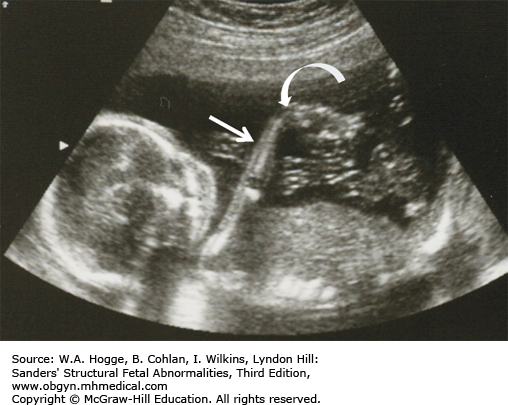
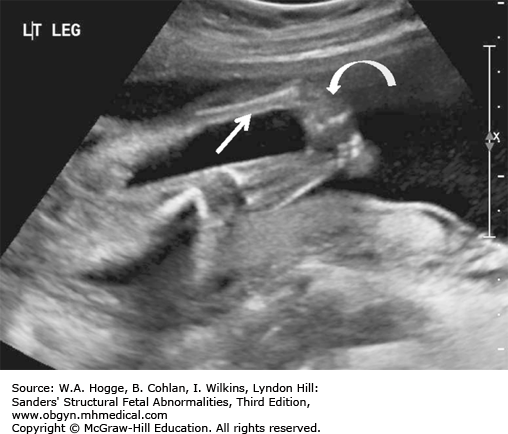
Definition Campomelic dysplasia is a skeletal dysplasia usually characterized by bowing deformities of the femur and tibia.
Epidemiology Incidence: rare. Phenotypic sex ratio, M1:F3; karyotypic sex ratio, M1:F1.
Embryology Infants with campomelic dysplasia have a flat facial profile, cleft palate, and tracheal abnormalities. Other abnormalities that have been associated are cardiac and renal anomalies. There is a less-common form of the disorder, known as acampomelic campomelic dysplasia, that has milder skeletal abnormalities and lacks long-bone curvature. An apparent preponderance of females is due to 46,XY males with sex reversal. The gene associated with the disorder is located on chromosome 17 and is designated SOX-9 (SRY-related HMG-box 9) gene. Mutations in the gene are responsible for both forms of campomelic dysplasia.
Inheritance Patterns Inheritance is autosomal dominant, with all cases representing new mutations. Campomelic dysplasia is due to mutations in the SOX9 gene on chromosome 17q. Recurrence in families has been reported on the basis of both somatic mutations and germ cell mutations in the parents. Consultation with a geneticist or genetic counselor concerning antenatal testing should be done in suspected cases.
Teratogens None are known.
Prognosis Usually, this is lethal in the neonatal period. Survival duration is related to the degree of respiratory compromise at birth. Infants with less-severe manifestations may occasionally survive into early infancy and beyond, but with significant respiratory and feeding difficulties. All have severe intellectual disability.
Extremities
Moderate to markedly shortened femur and tibia
Anteriorly bowed femur and/or tibia
Skin dimpling over the convex surface of the bone at the point of maximum deformity
Abnormal ratio of upper to lower limb
Upper limbs normal to shortened 25% versus 50% to 70% shortening of the lower limbs
Upper long bones occasionally bowed
Normal bone echogenicity
Hypoplastic/absent fibula
Hypoplastic scapula
Clubfeet
Thorax
Bell shaped
Mild narrowing
Eleven pairs of ribs
Face
Protuberant forehead
Micrognathia
Flattened nasal bridge
Low-set ears
Genitourinary
Ambiguous genitalia
Polyhydramnios
Upper long bones bowed
Hypotelorism
Cleft palate
Cloverleaf skull
Ventriculomegaly
Pyelocaliectasis
Ventricular septal defect
Aortic stenosis
Kyphoscoliosis
Three-dimensional imaging provides additional information that assists in the diagnosis of campomelic dysplasia. For example, 3-D skeletal mode can be used to evaluate the fetus for the 11 pairs of ribs associated with campomelic dysplasia.
Investigations and Consultations Required There are two goals of further investigations: determining whether the skeletal dysplasia is lethal and, if possible, determining which skeletal dysplasia the case represents. Campomelic dysplasia is particularly difficult, as some cases have been survivors, but most are lethal. If the precise skeletal dysplasia is uncertain, other investigations are warranted, including a fetal echocardiogram. Congenital heart disease is a rare feature of campomelic dysplasia. As gender may be phenotypically reversed, a karyotype is a useful adjunct to determining the diagnosis of campomelic dysplasia. Molecular testing on amniocytes can be done if there is a high suspicion of campomelic dysplasia. Once a diagnosis is established, and if the pregnancy is beyond the legal limits for termination, the family should meet with a neonatologist to discuss neonatal management. Consultation with a pediatric dysmorphologist may be beneficial to the family to discuss the natural history of those infants who survive.
Monitoring If the diagnosis is uncertain, follow-up ultrasound examinations may be helpful to look for features of the bones, such as the degree of bowing and the number of ribs, which may help to determine the diagnosis. Once the diagnosis is made, ultrasound examinations may be helpful to monitor facial features, such as a small chin, which may affect airway management at birth. Chest size is also a predictor of lethality and may be useful to monitor.
Pregnancy Course Mild polyhydramnios may develop, but preterm labor or other obstetric complications are unusual.
Pregnancy Termination Issues An intact fetus must be delivered, and the postmortem examination, including radiologic, morphologic, biochemical, and molecular studies, must be performed by individuals with extensive experience in skeletal dysplasias. Tissue for DNA testing should be obtained.
Delivery The site for delivery should be one where all personnel are comfortable with a nonaggressive approach to labor, delivery, and neonatal care, if elected by the family. Respiratory complications are common, and intubation may be complicated because of micrognathia. Delivery should occur in a tertiary center.
Resuscitation When the diagnosis is known prior to delivery, a prenatal decision for nonintervention, made after discussion with the family, is appropriate. If the diagnosis is uncertain, resuscitation and ventilatory support are instituted to allow time for diagnostic evaluation and parental adaptation.
Transport Referral to a tertiary perinatal center is appropriate for confirmation of a diagnosis. Mechanical ventilatory support is often required during the transport.
Testing and Confirmation Skeletal radiographs postnatally will establish the diagnosis.
Nursery Management Confirmation of the diagnosis and counseling and support of the family are the primary goals. Provision of mechanical life support initially is appropriate, with concurrence of the family. Redirection of care with comfort measures only may be offered if severe respiratory insufficiency persists.
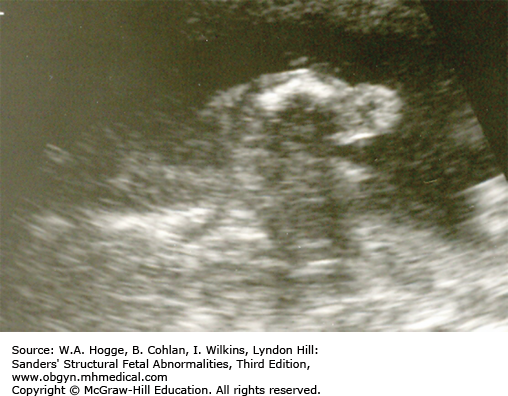
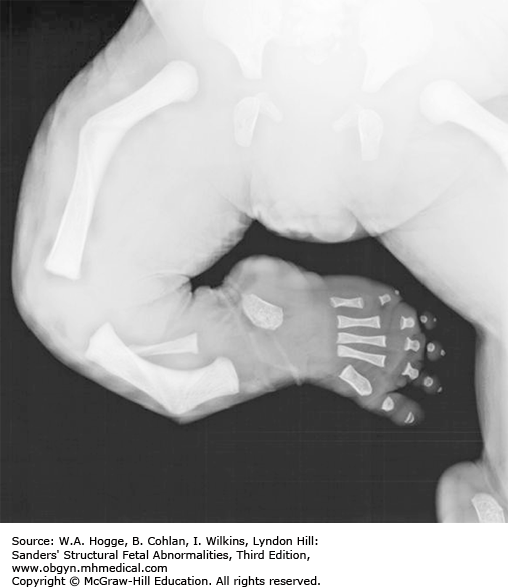
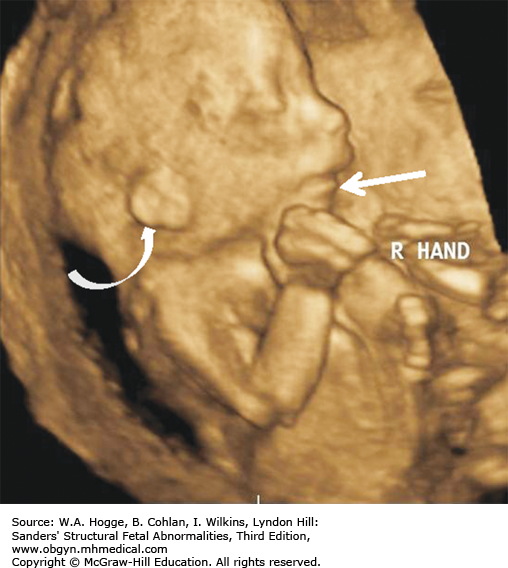
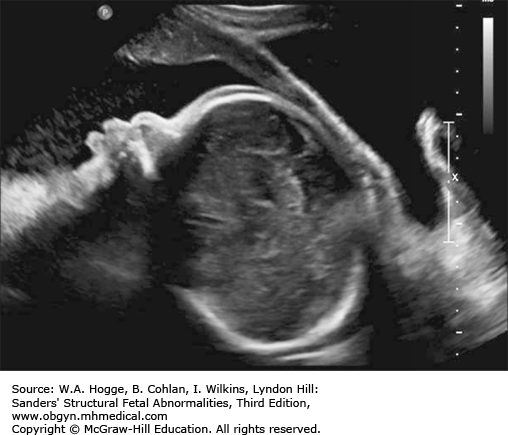
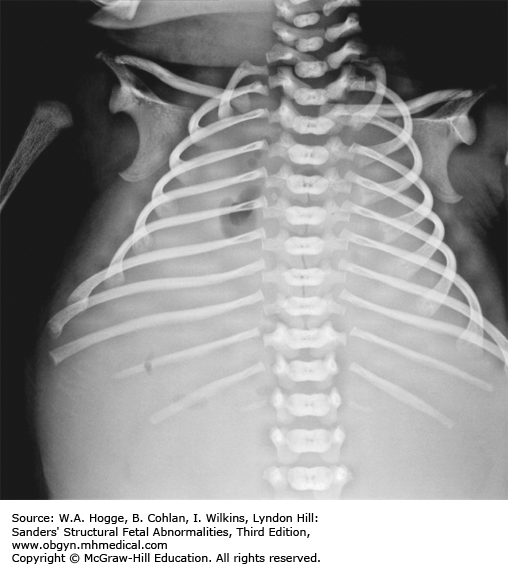
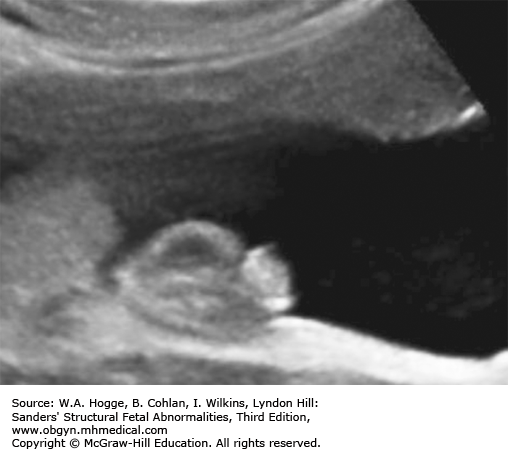
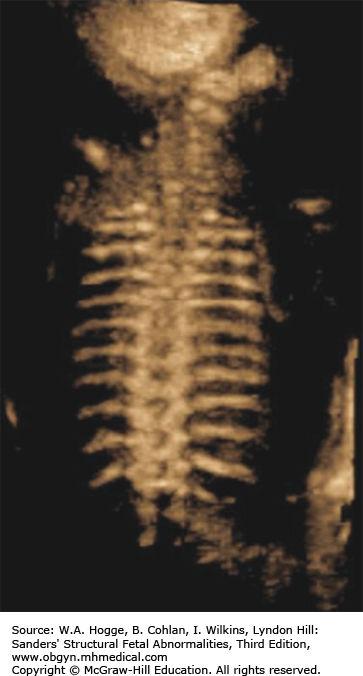
Definition Clubfoot is characterized by abnormal bone formation in the foot. There are four variations of clubfoot, including talipes varus, talipes valgus, talipes equinus, and talipes calcaneus. In talipes varus, the most common form of clubfoot, the foot generally turns inward. In talipes valgus, the foot rotates outward. In talipes equinus, the foot points downward. In talipes calcaneus, the foot points upward, with the heel pointing down.
A rocker-bottom foot is the phenotypic description of a foot characterized by dorsiflexion of the forefoot, equinus of the heel, and a convex sole.
Epidemiology Clubfoot occurs with a frequency of 1 to 3 in 1000 live births (M2:F1), making it the most common musculoskeletal abnormality. The frequency of rocker-bottom feet is unknown, but it is much less common.
Embryology Clubfoot can occur as an isolated abnormality or as part of more than 200 chromosomal, genetic, or sporadic multiple-malformation syndromes. The incidence of associated abnormalities has been reported to be between 24% and 50%. It is frequently secondary to lower extremity paralysis from a neural tube defect. There are also extrinsic causes of clubfoot, such as oligohydramnios, uterine cavity abnormality (leiomyomas), and amniotic bands. Rocker-bottom feet have been described in association with more than 30 chromosomal, genetic, or sporadic multiple-malformation syndromes, especially trisomy 18. Other more common causes of rocker-bottom feet are arthrogryposis syndromes and the multiple pterygium syndrome.
Inheritance Patterns Inheritance is dependent on the cause of the foot abnormalities.
Teratogens None are reported.
Prognosis Prognosis is dependent on the cause of the foot abnormalities. The outcome of an isolated clubfoot is excellent.
Tibia and fibula visualized with the lateral aspect of the foot.
The foot is flexed or extended and fixed in position.
Recheck the foot on a second scan to ensure it is not positioned to give the appearance of a clubfoot.
When a clubfoot is detected, the fetus should be evaluated for other congenital anomalies/syndromes. In many cases of suspected isolated clubfoot, associated anomalies may be undetected prenatally.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


