Definition Anal atresia is the congenital absence of an anal opening.
Epidemiology Occurrence is 1 5000 births (M3:F2).
Embryology Anal atresia results from an arrest in the division of the cloaca into the urogenital sinus and rectum that occurs during the ninth week of fetal development. There are two types: (1) high anal atresia, which is above the levator sling and is associated with urinary tract fistula in males or complex cloacal anomalies in the female; and (2) low anal atresia, which is below levator sling and is not associated with urinary fistula in males or the vagina in females. More than 80 genetic, chromosomal, and sporadic syndromes have been reported with anal atresia. Associated abnormalities occur in 85% of cases and include spinal/skeletal abnormalities, genitourinary abnormalities (50%), tracheoesophageal abnormalities, and cardiac malformations. The VACTERL (vertebral defects, anal atresia, cardiac defects, tracheoesophageal fistula [TEF], renal anomalies, limb abnormalities) association should be considered in these cases. Caudal regression and the OEIS (omphalocele, exstrophy of the bladder, imperforate anus, and spinal defects) sequence also may have anal atresia as a feature. Trisomies 18 and 21 both have been reported in cases of anal atresia.
Inheritance Patterns Inheritance is sporadic, with a 3% to 4% recurrence risk in first-degree relatives. However, the genetic syndromes with anal atresia as a feature include ones that are autosomal recessive, autosomal dominant, and multifactorial.
Teratogens Alcohol, thalidomide, and maternal diabetes are teratogenic.
Prognosis Prognosis is dependent on associated malformations and the nature of the anal atresia. Of isolated cases, 80% to 90% will have successful functional repair.
Absence of an “anal dimple”
Dilated colon/rectum (15% to 40%)
Associated anomalies (85%)
Urogenital anomalies (54%)
Bilateral renal agenesis
Urethral atresia
Cloacal anomaly
Calcified meconium due to vesicocolic fistula
There is a low prenatal detection rate: sensitivity (8%); positive predictive value (15%).
First-trimester prenatal diagnosis has been reported.
Early bowel dilatation may resolve.
Primary diagnostic clue: enterolithiasis: Coprolites form in the intestine in the presence of urine.
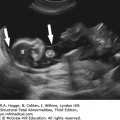
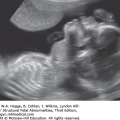
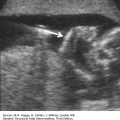
A V- or U-shape segment of dilated bowel in the lower pelvis suggests anorectal atresia.
The absence of the fetal anal sphincter can be used to diagnose an imperforate anus.
A transverse image of the anus has a “bull’s-eye” appearance.
Associated anomalies allow prenatal diagnosis.
Most frequently, it is diagnosed as part of VACTERL.
Anal atresia is associated with complete or partial urorectal septal malformation (URSM).
In complete URSM, there is an absence of the perineal and anal opening.
Usually, this is lethal due to pulmonary hypoplasia from oligohydramnios.
In partial URSM, there is a single outlet for urine and feces.
Dilated bowel may not be apparent until the third trimester.
Bowel dilatation may be transient in the second or third trimester.
Hirschsprung disease
There is congenital absence of ganglion cells of the mysenteric plexus.
There is a dilated loop of distal bowel.
Haustral markings are present.
There is an absence of peristalsis.
Bowel involvement:
Entire colon: 10%
Rectosigmoid: 74%
Distal-to-splenic flexure: 90%
Ten percent have associated meconium peritonitis.
Investigations and Consultations Required Fetal echocardiography should be performed to diagnose associated cardiovascular anomalies. Amniocentesis should be performed for chromosomal analysis and chromosomal microarray, especially if other anomalies are present. Fetal magnetic resonance imaging (MRI) may be helpful in defining the type of lesion and presence of associated anomalies. Consultation with a pediatric surgeon is indicated.
Monitoring Standard obstetric care is appropriate.
Pregnancy Course No specific obstetric complications are to be expected.
Pregnancy Termination Issues The late clinical presentation of this malformation generally will preclude the option of pregnancy termination.
Delivery The high association with other abnormalities makes delivery at a tertiary center with multiple pediatric and surgical subspecialists the best option. A pediatric geneticist should be available to evaluate the neonate for a possible genetic syndrome.
Resuscitation There are no special issues for delivery room management with isolated anal atresia. If other associated anomalies exist, then management may need to be directed toward their related issues.
Transport Transfer to a tertiary center having a pediatric surgeon is always indicated. Beyond orogastric decompression, there are no special precautions during transfer.
Testing and Confirmation This should be obvious on newborn examination. Postnatal radiologic studies will define the defect. The newborn should undergo plain films of the lumbar-sacral segments to assess for completeness, as this will have a significant impact on the long-term prognosis regarding continence. In addition, a spinal ultrasound should be completed for assessment of a tethered cord, a commonly associated finding. Renal ultrasound and cardiac echocardiogram are done to complete the workup and assess for other anomalies.
Nursery Management Once the lack of patency of the anus has been established, all enteral intake is contraindicated. Maintenance intravenous fluids should be administered. Nasogastric decompression should be maintained.







Definition Duodenal atresia is characterized by complete obliteration of the lumen of the duodenum and is the most common type of congenital small bowel atresia.
Epidemiology Incidence is 1 in 6000 to 1 in 10,000 births or 1 in 3500 in the second trimester.
Embryology At 6 weeks of embryonic life, the lumen of the duodenum is obliterated by proliferating epithelium. Patency is usually restored by the 11th week. Failure of this recanalization results in duodenal atresia. Another anatomical mechanism causing duodenal obstruction is an annular pancreas. An annular pancreas most likely results from a failure of completion of pancreatic rotation, and as a result, there is pancreatic tissue present circumferentially around the duodenum. Associated abnormalities occur in 50% of patients, with trisomy 21 occurring in 30% to 40% of cases. Additional abnormalities include skeletal defects (vertebral, rib, sacral agenesis, radial anomalies, and clubfoot); gastrointestinal abnormalities (annular pancreas, esophageal atresia [EA], TEF, intestinal malrotation, Meckel’s diverticulum, and anal atresia); cardiac malformations; and renal defects. More than 15 genetic, chromosome, and sporadic syndromes have been reported with duodenal atresia.
Inheritance Patterns Generally, inheritance is sporadic, with rare reports of autosomal dominant inheritance.
Teratogens Maternal diabetes is teratogenic.
Prognosis Mortality is due to associated abnormalities or preterm labor (50% of cases). With current neonatal and surgical management, mortality and morbidity are only about 5%.
Distended stomach
Distended duodenum
Continuity between a distended stomach and duodenum
Associated congenital anomalies
Trisomy 21
Cardiac
Tracheoesophageal fistula
Renal anomalies
Bowel atresias
Vertebral anomalies
Late second- and third-trimester polyhydramnios
Duodenal duplication cyst:
There is lack of communication between a dilated stomach and the duplication cyst.
Earliest diagnosis of duodenal atresia is at 14 weeks.
Usually, it is identified after 24 weeks’ gestation.
Gastric motility and emptying increase significantly after 24 weeks’ gestation.
Duodenal atresia is occasionally associated with EA, resulting in a “C-shaped” upper abdominal cystic mass. The combination of EA with TEF and duodenal atresia may result in surgical urgency or emergency due to the increasing distension of the obstructed stomach.
Investigations and Consultation Required An extended ultrasound examination to search for any anomalies in other organ systems should be undertaken. A fetal echocardiogram is warranted to exclude fetal heart defects, as these are relatively common. A fetal karyotype and microarray should be performed. Consultation with neonatology and a pediatric surgeon is indicated for the family to discuss neonatal management.
Fetal Intervention No intervention is warranted.
Monitoring The diagnosis is usually made in the third trimester. Follow-up ultrasound examinations to monitor the degree of polyhydramnios should be scheduled monthly, depending on maternal symptoms. As preterm labor is common, prenatal visits should also be scheduled at intervals to detect this and the patient educated on signs and symptoms.
Pregnancy Course The major obstetric complication of this disorder is severe polyhydramnios with a high incidence of preterm labor. In cases of polyhydramnios with severe maternal discomfort or respiratory compromise, a therapeutic amniocentesis is indicated. Whether it also abates preterm labor is less certain. As the fluid will reaccumulate rapidly, this procedure may need to be repeated.
Pregnancy Termination Issues Pregnancy termination is rarely an issue as the options for termination are limited by the late clinical presentation of duodenal atresia.
Delivery The site for delivery should be one capable of managing a preterm infant and one where appropriate personnel are available for surgical repair.
Resuscitation With high obstruction, amniotic fluid may be green stained from regurgitated bilious secretions, giving the erroneous impression that there has been meconium passage in utero. Aspiration of bilious material into the lungs may be damaging, and every effort should be made to prevent its occurrence. If there are indications of respiratory distress, it is safer to assume that aspiration may have occurred, and laryngoscopy and tracheal suction are indicated before the onset of respiration. In the absence of other markers of fetal distress, and if the fluid is thin and without particulate matter, airway instrumentation is probably not indicated. If positive-pressure ventilation is required because of respiratory depression, bag and mask ventilation should be avoided to reduce the likelihood of gaseous distension of the stomach and duodenum.
A nasogastric tube should be placed and the stomach emptied prior to transfer of the infant from the delivery room to reduce the risk of regurgitation and aspiration.
Transport Transfer to a tertiary center where there is a pediatric surgeon is always indicated. Orogastric decompression and maintenance of intravenous fluid infusion in transit are essential.
Testing and Confirmation Confirmation of the diagnosis is best established by abdominal radiographs or abdominal ultrasound. The classic radiograph shows the double bubble of an air-filled stomach and proximal duodenum. Air is an excellent and safe contrast medium for suspected upper tract obstruction. Contrast studies may be indicated if the patient is stable and other causes of duodenal obstruction are suspected, such as malrotation with volvulus. Although duodenal atresia may be an isolated finding, it can be associated with other anatomic malformations or chromosomal anomalies. Additional diagnostic evaluation for associated cardiac, gastrointestinal, renal, and vertebral anomalies should be obtained, as indicated. If trisomy 21 is suspected or diagnosed antenatally, confirmatory chromosomal analysis should be obtained at the time of delivery.
Nursery Management If cardiorespiratory resuscitation has been required, the first priority is to facilitate cardiorespiratory adaptation with oxygen, fluids, and ventilatory support as needed. Intravenous access should be established and fluid and electrolyte support instituted.
Nasogastric decompression should be maintained to minimize the risk of aspiration and respiratory complication. Parenteral nutrition is often required in the postoperative period, as there is usually a significant interval before adequate enteral intake is established.
Preoperative Assessment Most neonates present with an antenatal diagnosis of duodenal atresia, allowing for a thoughtful preoperative evaluation and elective scheduling of the operative repair. The most critical aspect of preoperative evaluation is the assessment for the presence of complex congenital heart disease. An echocardiogram should be completed preoperatively and findings of significant congenital heart disease discussed with cardiology and anesthesia to achieve consensus regarding a safe operative plan. Surgery may be deferred in the neonate born prematurely to allow for adequate weight and overall maturation prior to operative intervention. It is critical that the neonate has had adequate fluid and electrolyte resuscitation prior to surgical repair.
Operative Indications The surgical management of the duodenal atresia is relatively elective. That is, the operation can be safely delayed while possible associated anomalies are evaluated and the patient is stabilized. The exception to this is the neonate with both tracheoesophageal fistula and esophageal atresia (TEF/EA) and duodenal atresia. In this situation, the stomach may rapidly become distended, and even perforate, due to the distal obstruction. Depending on the neonate’s hemodynamic stability and respiratory status, a staged operative approach may be needed to allow adequate decompression of the obstructed stomach with eventual repair of all associated anomalies. When the neonate’s status allows, repair of the TEF/EA, allowing nasogastric decompression of the stomach prior to repair of the duodenal atresia, is generally optimal.
Types of Procedures Nasogastric decompression is immediately instituted, as well as intravenous fluid management. The defect usually occurs at the level of the second to third portion of the duodenum and frequently is in close association with the ampulla and the sphincter of Oddi. Most commonly, the obstruction is proximal, or cephalad, to the ampulla, resulting in nonbilious nasogastric drainage. The operation of choice is a duodenoduodenostomy, with careful evaluation at the time of laparotomy, or laparoscopy, to assess for intestinal malrotation or other sites of bowel atresia. The dilated proximal duodenum can have poor peristalsis with slow emptying across the anastomotic site, making delayed enteral nutrition a possibility. Total parenteral nutrition via a central venous line may be required, or a transanastomotic feeding nasojejunal tube may be used for earlier enteral nutrition. In neonates with Down syndrome, delay in return of gastrointestinal function should prompt an evaluation for Hirschsprung disease.
Surgical Results/Prognosis A major associated, and potentially lethal, problem is the presence of a cardiac defect in 20% of patients, which accounts for high mortality and chronic complication rates. Uncomplicated duodenal atresia has an excellent prognosis, with a greater than 95% survival rate.




Definition Gastroschisis is the intrauterine evisceration of fetal intestine through a paraumbilical abdominal wall defect. No covering membrane is present.
Epidemiology Incidence is 1 in 2000 births (M1:F1). There is an increased rate in women under the age of 20 years and those identifying as Caucasian. Increased incidence worldwide over the last 35 years suggests an environmental effect.
Embryology There are multiple theories of pathogenesis, including
Abnormal regression of the right umbilical vein
Vascular accident
Abnormal folding of the body wall
Developmental disruption of the right omphalomesenteric artery
The cause is probably heterogeneous, with some cases of gastroschisis resulting from vascular accidents and some from errors in development. Thickening, edema, and matting together of the intestines occur in some cases. Originally, it was theorized that these abnormalities were due to the irritating effects of amniotic fluid, but they are more likely the result of vascular compromise of the blood vessels coming through the small paraumbilical defect. Intestinal atresias and other gastrointestinal disruptions are found in approximately 5% to 10% of cases. Malrotation is almost universal. Extraintestinal abnormalities occur in 5% to 15% of cases. Ten to 15% of affected infants have long-term developmental disabilities, most likely secondary to the effects of prematurity. Both prematurity and intrauterine growth restriction may be associated with gastroschisis. Approximately 30% of fetuses with gastroschisis are delivered prior to 37 weeks’ gestation, and 30% to 50% are found to have intrauterine growth restriction.
Inheritance Patterns Rare familial recurrences suggest that a few families may have an autosomal dominant inheritance of isolated gastroschisis.
Screening Maternal serum α-fetoprotein measurement (MSAFP) will detect most gastroschisis cases. The median maternal serum α-fetoprotein level is 7.0 multiples of the median (MOM) for gastroschisis. With MSAFP and ultrasound, 90% to 97% of cases are detected prenatally.
Teratogens There is an association with maternal smoking and cocaine use, but it is not known if they are teratogenic or, more likely, are markers of other potential etiologies, such as poor diet.
Prognosis Mortality is 5% in utero, with most losses occurring before 36 weeks. There is less than a 10% neonatal death rate in the postoperative period due to complications of surgery or bowel atresia.
Abdominal wall defect to the right of the umbilical cord insertion
Multiple loops of small bowel within the amniotic cavity: “cauliflower” appearance
Large bowel in the amniotic cavity
Stomach pulled toward the abdominal defect
Distended fetal stomach within the abdomen
Intra-abdominal small bowel atresias
Abnormalities of amniotic fluid volume
Oligohydramnios
Polyhydramnios
The abdominal wall defect in gastroschisis is usually less than 20 mm.
Intra-abdominal bowel dilatation in the third trimester suggests an obstruction.
Vanishing gut involves complete necrosis of an affected loop of bowel and closure of the abdominal wall defect.
Gastroschisis is associated with volvulus and bowel atresias.
Bowel dilatation does not mandate early delivery.
There is an increased risk of prematurity and intrauterine growth restriction (40%).
Polyhydramnios may be associated with severe bowel complications.
The fibrous peel on the intestines develops after 30 weeks’ gestation.
Physiologic gut herniation occurs up to 12 weeks’ gestation.
Physiologic herniation is into the base of the umbilical cord, permitting its differentiation from gastroschisis.
Omphalocele:
Characteristically, this is a larger abdominal wall defect containing liver.
Bowel-containing omphaloceles can only be diagnosed after 12 weeks’ gestation because of the physiologic herniation of bowel into the base of the umbilical cord.
Investigations and Consultations Required An extended sonographic examination should be performed to exclude other anomalies and to characterize the pattern of bowel findings. The fetal size should be recorded. Fetal echocardiography should be performed. Although the risk of chromosomal abnormalities is low, amniocentesis should be discussed with the patient. If performed, or if other fetal anomalies are present, a microarray is indicated. The family should be referred to neonatology and a pediatric surgeon for a thorough discussion of the neonatal management issues.
Monitoring Care should be coordinated in a tertiary center because of the potential need for early intervention. Serial ultrasonographic examinations should be performed every 4 weeks to detect thickening or dilation of the fetal bowel, and to assess fetal growth and amniotic fluid abnormalities. If abnormalities develop, ultrasound examinations may be performed more frequently. Fetal assessment, such as biophysical profiles, should be initiated if there is evidence of a lag in growth or if oligohydramnios is present.
Pregnancy Course The increased incidence of intrauterine growth restriction in fetuses with gastroschisis may complicate the obstetric management. Preterm labor occurs in nearly one-third of cases.
Pregnancy Termination Issues There are no special concerns regarding the method or location for pregnancy termination, as the diagnosis is generally clear antenatally.
Delivery Delivery at term is preferred, when fetal growth and amniotic fluid are reassuring. Delivery prior to 39 weeks is based on the usual obstetrical concerns for fetal growth restriction and nonreassuring antenatal testing. Vaginal delivery is appropriate with care to immediately cover the protruding bowel loops with non-latex-containing material. Delivery personnel should wear non-latex-containing gloves and carefully identify where to clamp and cut the umbilical cord. A pediatric surgeon and a tertiary neonatal intensive care unit should be available at the delivery site.
Resuscitation Assistance with the onset of respiration is usually not required unless there is concurrent prematurity-associated respiratory distress. If assisted ventilation is required, bag-and-mask ventilation is contraindicated to avoid gaseous distension of the stomach and bowel. The major concern is the protection of the extruded bowel. Extreme care must be taken to avoid torsion of the bowel loops, which would further compromise perfusion. If there is marked distension of the bowel, perfusion can be compromised by the kinking of the mesenteric vessels as they exit through the abdominal wall defect. Prompt decompression of the stomach is important. The bowel should be covered with warmed saline-soaked gauze and supported to avoid torsion, and the trunk should be encased in a sterile plastic bag with a drawstring (“bowel bag”). The bowel bag not only reduces evaporative heat and water loss but also reduces the likelihood of surface contamination. The neonate should be positioned laterally to minimize torsion on the bowel and its mesentery, which could occur with the neonate positioned supine.
Transport Transfer to a tertiary center with pediatric surgery and neonatology is always indicated. Care of the bowel, as outlined, should be provided during the transport, as should gastric decompression. Reliable venous access should be established, and maintenance intravenous fluids and broad-spectrum antibiotic therapy should be instituted.
Testing and Confirmation Once the infant has recovered from the initial surgery and the exposed bowel is covered, additional diagnostic evaluation may be instituted if there is concern for associated anomalies.
Nursery Management Protection of the exposed bowel, gastric decompression, administration of broad-spectrum antibiotics, and intravenous fluid support should be maintained. In some instances, it may be necessary to provide ventilatory support in the early postoperative period, as there may be increased intra-abdominal pressure from the reduction of the extruded bowel back into the abdominal cavity. Central parenteral nutrition is required during the postoperative recovery period, as there is always a significant interval before adequate enteral intake is established.
Surgical Management The goal of surgical management is to reduce the eviscerated bowel into the abdominal cavity, a task complicated by the common finding of a small abdominal cavity, due to intrauterine loss of domain, and the presence of thickened bowel loops. The decision to proceed with a staged, rather than a primary, closure is dependent on the findings at the time of operative repair, specifically increased intra-abdominal pressure and its effects on ventilation and the perfusion of the intestines. A preformed, spring-loaded silo is most commonly used to protect the bowel in a staged closure, and these silos may be placed without general anesthesia, allowing placement in the neonatal intensive care unit, with gradual reduction of the intestines into the abdominal cavity over several days. A suture-less closure using the umbilical cord is a relatively new, innovative approach to the abdominal wall closure in gastroschisis. With this technique, the umbilical cord is used to plug the fascial defect with relatively rapid epithelialization of the wound. This results in a small hernia at the umbilicus that often closes with time.






Definition Liver tumors are the tenth most frequent pediatric tumor. Hepatic malignancy must be distinguished from benign hepatic tumors and nonneoplastic hepatomegaly. Almost 50% of primary liver tumors are benign. In older children, liver tumors more often represent metastatic disease (neuroblastoma, Wilms tumor) than primary malignancies.
Epidemiology Occurrence is 1.6 in 1 million births; for hepatoblastoma, incidence is M1.4:F1, and for vascular tumors it is M:1.3:F1.
Embryology There are three major types of hepatic tumors with the following characteristics:
Hepatoblastomas (17%)
Hepatoblastomas present as an abdominal mass, with 60% detected in the first year and 90% by age 3 years.
They occur most frequently in the right lobe of the liver.
They are usually unifocal.
Hepatomas are locally invasive.
They can metastasize.
α-Fetoprotein is elevated.
The presence of embryonal or undifferentiated cell types is associated with poor prognosis.
Punctate calcifications may be present.
There is an association with the Beckwith-Weidemann syndrome, hemihypertrophy, diaphragmatic hernia, and renal abnormalities.
Mesenchymal hamartomas (23%)
These are benign.
Etiology: They arise from an overgrowth of mature cells.
They can be solid, large septated, or unilocular cysts filled with mucoid material.
Rapid growth can lead to hydrops.
Hemangioma (60%).
Types
Capillary and cavernous hemangioma
Hemangioepithelioma
Infantile hemangioma
Can be focal and multifocal lesions
Can be self-involuting
May cause consumptive coagulopathy
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


