vascular scalp lesions, color and duplex Doppler are added to demonstrate lesional vascularity and pulsatility of blood flow (i.e., arterial versus venous).
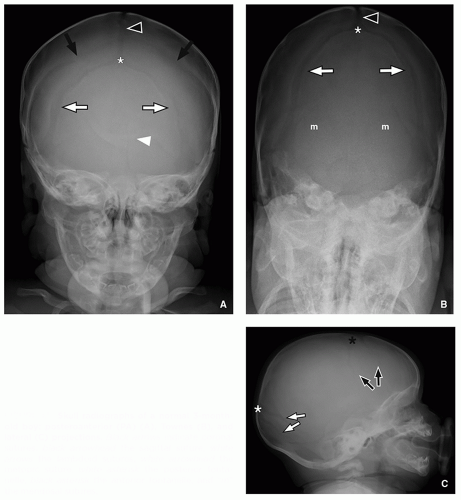 FIGURE 1.1 Skull radiographs of a normal 3-monthold boy: posteroanterior (PA) (A), Townes (B), and lateral (C) projections. Black arrows indicate coronal sutures, black arrowhead the sagittal suture, white arrows the lambdoid sutures, white arrowhead the metopic suture, white asterisk the posterior fontanelle, black asterisk the anterior fontanelle, and “m” the mendosal sutures. |
create multiplanar reformatted images and surface rendered data, the latter particularly helpful in delineating complex 3D relationships of skull lesions. In our institution, an agespecific dosage scheme17 compliant with recommendations from both the Society for Pediatric Radiology Image Gently campaign and the ACR CT Accreditation Requirements is used,18,19 the latter limiting the CT dose index (CTDIvol) for a 1-year-old to 40 mGy compared to 80 mGy for an adult. For pediatric patients undergoing evaluation for craniofacial dysmorphism, the field of view is extended below the mandible to capture any facial bone abnormalities, and the dose is reduced to 100 kV and 50 mAs, resulting in a CTDI <5 mGy. The trade-off for this reduced dose is poorer parenchymal detail. Even with these attempts to reduce dose, it is worth noting that a standard head CT still carries an estimated dose that is 20 times a single projection skull radiograph.20
sutures and skull base synchondroses are patent at birth and then begin to close at variable rates. However, two useful rules of thumb are that no major suture should close in the first year of life and no suture should undergo mature fusion in childhood except for the metopic suture, which normally closes at 3 to 9 months.44,50,51,52,53,54 As the patient ages, the sutures become more serrated at their outer table though they remain smooth along the inner table.55,56 For cases where there is subjective narrowing, normative neonatal data have been published for selected sutures based on CT data.57 Important synchondrosis milestones include closure of frontosphenoid and intersphenoid synchondroses by 1 to 2 years of age and of the sphenoccipital (clival) synchondroses during adolescence.58 Closure of synchondroses and sutures within the occipital bone follow a more complicated sequence. The synchondroses at the foramen magnum typically close within the first few months of life though other sutures including mendosal sutures and inferior occipital fissure may persist through the first few years of life47,48,58,59; variants where these sutures/synchondroses persist into adulthood are occasionally encountered. The anterior and posterolateral fontanelles typically close by the end of the 2nd year. The posterior fontanelle closes much earlier, typically by 3 to 6 months.60
 FIGURE 1.2 Three-dimensional surface renderings of a head CT from a normal 3-month-old boy evaluated for craniosynostosis: frontal (A), lateral (B), posterior (C), and superior (D) views. Annotations as for Figure 1.1 with additional markings of pterion with a circle, squamous (temporoparietal) suture with “s,” sphenotemporal suture with white dashed arrow, frontosphenoid suture with black dashed arrow, occipitomastoid suture with “o,” and posterior intraoccipital synchondrosis with “p.” |
mineralization (osteogenesis imperfecta, rickets, hypothyroidism) and can be misinterpreted as sutural diastasis from elevated intracranial pressure. Other reported causes of apparent widening of normal sutural width include recovery from chronic malnutrition, in utero renin-angiotensin system disruption (Fig. 1.4), achondroplasia, trisomy 21, and treatment with prostaglandins for prematurity.61,62,63,64,65
 FIGURE 1.3 Transient sutures of the occipital bone visualized on a three-dimensional surface rendering of a 1-monthold boy’s head CT (posterior view). Markings are as for Figures 1.1 and 1.2 but additional sutures marked include “so”, “m”, and “io” for superior occipital, paired mendosal sutures, and inferior occipital sutures, respectively. |
 FIGURE 1.4 Markedly widened cranial sutures (hypocalvarium) in a 1-day-old girl with renal tubular agenesis, pulmonary hypoplasia, and other skeletal anomalies suggesting in utero exposure to renin-angiotensin system blockers or an inborn error in the renin-angiotensin system. Reflection of the scalp at autopsy shows only a thin membrane covering much of the cerebrum. In the right panel, the margins of the fontanelles are outlined in black ink. |
fact may be explained by the increasing detection of mutations responsible for syndromic synostosis in “nonsyndromic” synostosis patients, and therefore many authorities advise genetic screening in coronal synostosis patients.74,75,76
 FIGURE 1.5 Sagittal synostosis in a 3-month-old boy with dolichocephaly and midline osseous ridging. A: Coronal reformatted bone window CT image shows premature fusion of the sagittal suture accompanied by osseous ridging (asterisked black arrowhead). B-D: Frontal (B), superior (C), and right lateral (D) surface rendering CT images confirm the osseous ridging and depict the elongated AP dimension of the skull consistent with clinical impression of dolichocephaly. When accompanied by osseous ridging, dolichocephaly is called scaphocephaly, a characteristic appearance for sagittal synostosis. Note that the metopic suture has already undergone fusion without deformity, consistent with normal closure. |
 FIGURE 1.6 Unilateral coronal synostosis in a 4-month-old boy with abnormal head shape. A: Frontal projection CT topogram demonstrates elevation of the left orbital rim. B: Axial bone window CT image demonstrates premature fusion of the left coronal suture (abnormal side asterisked arrow). C and D: Three-dimensional surface rendering CT images in the frontal (C) and superior (D) perspective demonstrate left frontal plagiocephaly and right frontal bossing with retraction of the metopic suture to the left. Labeling conventions as for Figure 1.2 with abnormally fused left coronal suture denoted by an asterisked black arrow. |
classic findings of metopic synostosis include retraction of the supraorbital ridges medially, bossing of the parietal bones, hypoplastic ethmoid sinuses, and a W-shaped metopic notch of the endocranial surface of the fused metopic suture (Fig. 1.8).54,78,81
 FIGURE 1.7 Bicoronal synostosis in a 2-month-old boy with skull deformity. A: Frontal projection CT topogram shows bilateral harlequin deformities of the orbits. B: Axial bone window CT image demonstrates bilateral anterior plagiocephaly and premature fusion of both coronal sutures (asterisked black arrows with metopic suture denoted by white arrowhead). C and D: Surface rendering CT images confirm premature fusion and ridging of the coronal sutures with upward retraction of the orbits in the frontal view (C) as well as brachycephaly on the lateral view (D). |
 FIGURE 1.8 Metopic synostosis in a 24-day-old boy with trigonocephaly. A: Axial bone window CT image confirms trigonocephaly with prominent osseous ridge and metopic notch (asterisked white arrowhead) at the site of the prematurely fused metopic suture. B and C: Frontal and superior surface rendering CT images (B and C) show retraction of the medial orbital, hypotelorism, and the contour deformity of the skull. D: Sagittal reformatted CT image demonstrates an incidentally noted persistent craniopharyngeal canal (arrow) anterior to the normal spheno-occipital synchondrosis (arrowhead). |
massive brain injury.86 In both instances, the intracranial volume contracts, resulting in overlapping sutures and premature fusion with osseous ridging (Fig. 1.13). Other reported associations include underlying disorders of bone metabolism, such as rickets; bone dysplasias, such as achondroplasia; metabolic disorders, such as mucopolysaccharidoses; in utero compression; hematologic disorders, such as sickle cell and polycythemia; endocrine disorders, such as hyperthyroidism; and any cause of poor underlying brain growth.87,88,89,90,91,92,93,94,95,96
 FIGURE 1.9 Comparison of lambdoid synostosis and posterior deformational plagiocephaly. A—C: A 6-month-old boy with left posterior plagiocephaly due to left lambdoid synostosis. Axial bone window CT image (A) demonstrates fusion and osseous ridging of the left lambdoid suture (white arrows, abnormal side asterisked). Frontal and posterior surface rendering CT images (B and C) show the premature fusion of the left lambdoid suture and also suggest subtle frontal bossing and skewing of the lambda to the left. D—F: Comparison case of a 6-month-old boy with deformational left posterior plagiocephaly. Axial bone window CT image demonstrates patency of the lambdoid suture on the affected side (D), confirmed with surface renderings in the frontal and posterior perspective (E and F). |
from true metopic synostosis by the absence of trigonocephaly.99 Dolichocephaly refers to narrowing of the cranial vault, according to some authors defined as a cephalic index (width:length of skull) <0.75.100 Dolichocephaly differs from scaphocephaly in that the latter has premature fusion/ridging of the sagittal suture, whereas the former is typically caused by positioning in premature infants.78,100
 FIGURE 1.10 Newborn girl with turricephaly and syndactyly at birth, subsequently diagnosed with Apert syndrome. A: Axial bone window CT image demonstrates bilateral coronal and lambdoid suture synostosis (fused coronal sutures marked with asterisked black arrows and lambdoid sutures with asterisked white arrows). B and C: Frontal (B) and right lateral (C) perspective surface rendering CT images confirm the synostosis and demonstrate the “towering” appearance of the cranial vault. D: Soft tissue windowing of the lateral perspective surface rendered CT image depicts the resulting deformity as viewed externally. |
later surgery (better tolerance of blood loss).101 However, evidence of elevated intracranial pressure (e.g., papilledema or lacunar changes mentioned under the section “Variants”) is widely accepted as an indication for early surgery regardless of institutional preference, being uncommon in single-suture synostosis and present in the majority of patients with syndromic, multisuture synostosis.81,102 Furthermore, a significant minority of syndromic patients require management of additional complications such as communicating hydrocephalus or Chiari I malformations (Fig. 1.11E).103,104 The syndromic patients also have a high incidence of midface hypoplasia with associated complications of exorbitism/keratitis and respiratory compromise from the midface retrusion.102,105,106 For single-suture craniosynostosis, surgery generally includes radial and barrel stave osteotomies to remodel the cranial vault and orbitofrontal advancement to normalize the position of the bony orbits.81 Although simple excision of an abnormal suture has historically been avoided because of poor outcomes, newer endoscopic approaches have found success in patients <6 months when augmented by spacers and postoperative helmeting.107 In the case of syndromic/multisuture craniosynostosis, it is not uncommon for revision surgeries to be performed later in addition to multistage advancement of the hypoplastic midface through LeFort osteotomies.102
 FIGURE 1.11 Kleeblatschädel or “cloverleaf” skull deformity in a newborn boy with Pfeiffer syndrome and pansynostosis. A and B: Axial (A) and coronal reformatted (B) bone window CT images demonstrate constraint along all major (coronal, lambdoid, sagittal) and some minor (squamosal) sutures with bulging of the cranial vault between the abnormally fused sutures. Note also the profound exorbitism. C and D: Frontal (C) and right lateral (D) perspective surface renderings depict the distorted cranial vault in three dimensions as well as the markedly enlarged anterior fontanelle. E: Midsagittal T2-weighted MR image demonstrates associated Chiari I malformation (white arrow) and hydrocephalus (ballooning of the infundibular recess of the third ventricle). |
TABLE 1.1 Features of the Syndromic Craniosynostoses | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
 FIGURE 1.12 Excessive lacunar markings in a 2-year-old boy with multiple suture synostosis and sinus pericranii. A and B: AP (A) and lateral (B) skull radiographs demonstrate prominent lacunar changes of the skull and poor visibility of the major cranial sutures. C and D: Right lateral (C) and posterior (D) views of postcontrast surface rendering CT images demonstrate premature fusion of the sagittal suture (asterisked black arrowhead) and portions of the lambdoid/coronal sutures (asterisked white/black arrows). There is also a tuft of veins along the outer table from a sinus pericranii (circle). |
American) may represent risk factors.110 Some authors classify cephaloceles as a defect of neural tube closure, similar to anencephaly or myelomeningocele for the anterior and posterior neuropore, respectively. However, the recurrence rate in siblings and lack of demonstrated decrease with folate supplementation differentiate cephaloceles from classic neural tube disorders.111,112 Therefore, many authors instead speculate on a variety of other developmental and physical mechanisms to explain the occurrence of these skull defects.
 FIGURE 1.13 Secondary craniosynostosis in a 3-year-old boy with neonatal hypoxic ischemic injury. A: Left lateral perspective surface rendering CT image demonstrates sutural overlap and fusion. B: Axial bone window CT image shows diffuse encephalomalacia underlying the sutural fusion. |
cephalocele; caution is required when making this diagnosis in neonates as the cribriform plate does not normally ossify before 6 months.119 A naso-orbital cephalocele takes a similar course but interdigitates between the lacrimal bone and maxilla (frontal process). Nasofrontal cephaloceles occur at the primitive nasofrontal suture, known as the fonticulus frontalis.120,121 These sincipital cephaloceles typically present as nasal or orbital masses, frequently disrupting the lacrimal ducts and causing hypertelorism.108,118 In Southeast Asia, Central Africa, and certain parts of Russia, sincipital cephaloceles constitute the most common type of cephalocele.108 The spectrum of anterior cephaloceles is depicted in Figures 1.15, 1.16 and 1.17.
TABLE 1.2 Classification Scheme for Cephaloceles Based on Location | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
 FIGURE 1.14 Occipital cephalocele in a girl at 30 weeks gestational age and 3-months of age. A: Sagittal HASTE fetal MRI obtained at 30 weeks demonstrates a suboccipital cephalocele (arrow) with question of a tongue of cerebellar parenchyma extending to the defect. B and C: Sagittal (B) and axial (C) FIESTA postnatal MR images demonstrate herniation of the left cerebellar hemisphere through the osseous/dural defect (arrow). |
 FIGURE 1.15 Large interfrontal cephalocele and Tessier facial cleft in a 16-month-old girl. A and B: Axial (A) and sagittal reformatted (B) CT images demonstrate a large interfrontal cephalocele. The sagittal reformatted CT image demonstrates that the axis of herniation extends above the frontonasal suture (arrowhead). C and D: Frontal (C) and left lateral (D) perspective surface renderings of the CT data demonstrate the large frontal bone defect, the presence of the left paramedian Tessier facial cleft (black arrow), and some residual bone at the frontonasal suture (white arrow).
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|