Abstract
This chapter describes primary and secondary bone tumors affecting the skull base and provides important epidemiologic and imaging clues for the differential diagnosis. Except for bone metastases, bone tumors of the skull base are rare. Although the same diagnostic rules apply for bone tumors of flat bones seen elsewhere in the skeleton, radiologists tend to forget about them, leading to missed or delayed diagnosis. A simple classification of bone-forming tumors, cartilage-forming tumors, and tumors without bone or cartilaginous matrix (of vascular, hematopoietic, fibrous, and giant cell origin), together with patient’s age can be quite helpful in limiting the differential diagnosis. Moreover, imaging is quite accurate in differentiating benign from malignant tumors, the former featured by slow, geographic growth, with well-defined margins and thick, onion skin-like periosteal reaction, and the later by rapid, permeative/destructive growth, with ill-defined margins and spiculated, sunburst-like, periosteal reaction. The presence of multifocal bone lesions further drops down the diagnostic possibilities to metastatic disease, multiple myeloma, hematologic malignancies, brown tumors, and eosinophilic granulomas. CT, MRI, bone scintigraphy, and PET-CT all contribute to the final diagnosis, and the main imaging features of different benign and malignant bone tumors affecting the skull base are reviewed in detail.
Keywords
benign bone tumors, bone tumors, imaging bone tumors, malignant bone tumors, Skull base, skull base imaging
Introduction
Most tumors affecting the skull base result from hematogenous spread of primary malignancies outside the skull base (hematogenous bone metastases) or from direct invasion or perineural spread of neoplasms arising from neighboring structures of the suprahyoid neck. As the skull base provides a frontier between the intracranial compartment and the extracranial head and neck, the first and most important issue in the differential diagnosis of a skull base lesion is to decide its site of origin: the bone elements of the skull base proper, the intracranial compartment, or the suprahyoid neck.
With the exception of metastases, bone tumors of the skull base are overall rare and can be a diagnostic dilemma. When facing a bone tumor in the skull base, the same rules used for the differential diagnosis of skeletal tumors elsewhere in the body should be applied, taking into account the specificities of this anatomic location regarding tumor extent and treatment planning. Although imaging features can be quite helpful in the differential diagnosis, they are not very familiar to radiologists who do not routinely deal with skull base lesions and only a scant specific literature is available on this subject. This chapter specifically focuses on primary and secondary skull base bone tumors. For the imaging technique, general rules for the differential diagnosis, and developmental and diffuse skull base lesions, please refer to the previous chapter ( Table 16.1 ).
| Bone Tumors | |
|---|---|
| Primary | |
| Benign | Osteoma |
| Osteoid osteoma | |
| Osteoblastoma | |
| Ossifying fibroma | |
| Enchondroma/osteochondroma | |
| Chondroblastoma | |
| Chondromyxoid fibroma | |
| Intraosseous hemangioma | |
| Aneurysmal bone cyst | |
| Giant cell tumor/osteoclastoma | |
| Brown tumor | |
| Eosinophilic granuloma | |
| Malignant | Metastasis |
| Plasmacytoma/multiple myeloma | |
| Lymphoma | |
| Chloroma/granulocytic sarcoma | |
| Ewing sarcoma | |
| Osteosarcoma | |
| Chondrosarcoma | |
| Secondary | |
| Metastasis | |
| Lymphoma | |
| Leukemia (chloroma) | |
| Diffuse Bone Lesions | |
| Fibrous dysplasia | |
| Paget disease | |
| Osteopetrosis | |
| Osteopoikilosis | |
| Melorheostosis | |
| Infection (skull base osteomyelitis) | |
| Multifocal Bone Lesions | |
| Metastases | |
| Multiple myeloma | |
| Brown tumors | |
| Eosinophilic granulomas | |
Bone Tumors
Primary bone tumors encompass a variety of benign and malignant neoplasms. With the exception of osteomas and intraosseous hemangiomas in the benign category and metastases and multiple myeloma on the malignant side, other tumors are quite rare in the skull base. The imaging features of these primary and secondary bone tumors are similar to those of bone tumors arising in other sites of the skeleton, although the treatment and implications on patient’s management derive from the specificities of this particular location. The imaging features of these lesions in the skull base are reviewed.
Benign Tumors
These can be divided into bone-forming lesions, characterized by the presence of a variable amount of ossified osteoid matrix (such as osteoma, osteoid osteoma, osteoblastoma, and ossifying fibroma); cartilage-producing tumors characterized by a variable amount of chondroid matrix and, commonly, by the presence of chondroid-like calcifications (such as enchondroma, chondroma, chondroblastoma, and chondromyxoid fibroma); and bone tumors that produce neither osteoid nor chondroid matrix, although they may contain fragments of remaining trabecular bone inside. The cellular component of these tumors is variable: osteoclast-like multinucleated giant cells, such as in giant cell lesions (giant cell tumor and brown tumors secondary to hyperparathyroidism); blood vessels and blood, such as in hemangiomas and in aneurysmal bone cysts; or abnormal proliferations of histiocytic cells, such as in eosinophilic granuloma. In this last category, most lesions present as lytic, punched-out, bubbly lesions with variable matrix depending on their cell of origin.
Bone-forming tumors
Although osteomas are rare, they are the most common benign primary skull base tumors, with an estimated prevalence of 0.4%–1%. Most cases are sporadic, but the association with Gardner syndrome is well documented. They can arise from any bone, although they are most often encountered in the anterior skull base arising in or around the sinonasal cavities. Their pathophysiology remains controversial. According to the embryologic theory, the most widely accepted, they arise from cartilaginous remnants in the junctional zone around the ethmoid labyrinth.
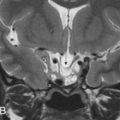
These osteogenic tumors can be of cancellous or compact bone, the latter also called ivory osteomas. They are often small lesions of no clinical significance, except for the potential obstruction of sinus ostia or infundibula with secondary development of mucoceles. Rarely, they can attain large dimensions, above 3 cm in size, and are then named giant osteomas. These lesions can become large enough to protrude into the anterior cranial fossa or orbital contents and become symptomatic, requiring surgical correction. On imaging, they are well-defined, sessile or pedunculated, sclerotic lesions, of compact or trabecular bone density/signal intensity ( Figs. 16.1 and 16.2 ).


Osteoid osteomas are also exceedingly rare in the skull base with only a few cases reported in the literature, most affecting the anterior skull base, in or around the frontal or ethmoid sinuses. Radiographically, these benign, slow-growing tumors are characterized by the presence of a radiolucent nidus, which may contain mineralized matrix, surrounded by reactive sclerosis. Large lesions, above 1.5 cm in size, are called giant osteoid osteoma or osteoblastoma and are featured by an increasing number of osteoclasts, a richly vascularized stroma, hemorrhage, and multinucleated giant cells responsible for a more heterogeneous imaging appearance with blood degradation products and enhancing septa. When prominent, the sclerotic component of an osteoid osteoma may obscure the radiolucent nidus and hamper the diagnosis. In these circumstances the main differential diagnosis is with sclerosing forms of osteomyelitis (Brodie abscess), ossifying fibroma, fibrous dysplasia, and osteogenic osteosarcoma. On bone scintigraphy (99mTc single-photon emission CT) the nidus of the lesion can be clearly identified as a “hot” zone, important information for surgery planning.
These tumors are usually seen in the second and third decades and are more common in males. Small lesions in the skull base are often asymptomatic, as pain is not a prominent feature in this particular location. Clinical symptoms most often result from compression or displacement of adjacent structures, depending on their primary location within the skull base.
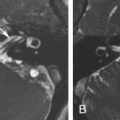
Osteoblastoma is a benign, osteoid-forming neoplasm, rich in osteoclasts, with a higher incidence in males, 90% occurring before 30 years of age and of rare occurrence in the skull base. They are considered osteoid osteomas larger than 1.5 cm in size presenting, on imaging, as expansive, sharply circumscribed lesions, predominantly lytic but often with a mixed lytic-sclerotic pattern reflecting different degrees of osteoid production and matrix mineralization. Postcontrast images typically show enhancement of the tumor stroma as multiple enhancing septa. The growth pattern is geographic, with a peripheral sclerotic rim resembling an egg shell and remodeling adjacent bone ( Fig. 16.3 ). The association with aneurysmal bone cysts is a common occurrence featured by cystic areas with fluid-fluid levels. Moreover, when large, areas of cortical disruption may ensue. Pathologically, two different forms of osteoblastoma have been recognized: benign and aggressive. The latter is an exceedingly rare, locally aggressive, and destructive tumor with intermediate histologic features between benign osteoblastoma and osteosarcoma. A review of 71 skull base osteoblastomas has shown that the most common location is the temporal bone (36%), followed by the frontal bone (18%). In this review, recurrence rates after surgery were 10% and 33% for gross total and subtotal resection, respectively, and higher, with an average of 57%, for the malignant counterpart. However, gross total resection may be difficult for large skull base lesions and is associated with high morbidity.

Ossifying fibroma is a benign, locally aggressive bone-forming tumor of young children, most commonly seen under age 10 years (juvenile ossifying fibroma). These tumors belong to the spectrum of fibroosseous lesions, which range from nonossifying fibromas (fibrous cortical defect) to fibrous dysplasia. Classification and nomenclature are confusing and a matter of controversy, including cementifying or cementoossifying fibroma, psammomatoid or juvenile ossifying fibroma, and ossifying fibromyxoid tumor. Some authors consider these as separate entities, whereas others believe they are variants of the same entity. To further complicate the matter, there are hybrid fibroosseous lesions containing elements of aneurysmal bone cyst, ossifying fibroma, and cementifying fibroma.
The conventional ossifying fibroma is a solitary, slow-growing lesion, which tends to be more aggressive in children. The jaws, skull base, and temporal bone are the most common locations in the craniofacial region. Lesions in the pediatric age group are subdivided into psammomatoid and trabecular ossifying fibromas, the former featured by the presence of psammoma body islands (juvenile psammomatoid ossifying fibroma) and the latter by a clinically aggressive behavior (juvenile active ossifying fibroma).
Clinical presentation depends on lesion location but is often by craniofacial deformity and compressive symptoms such as proptosis, nasal obstruction, and headache.
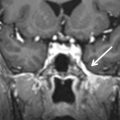
On plain films and CT imaging, ossifying fibroma presents as a well-circumscribed, often expansive, unilocular, osteolytic lesion with a sclerotic rim of variable thickness, reflecting an osteoblastic rimming, which may show a ground-glass appearance similar to fibrous dysplasia ( Figs. 16.4 and 16.5 ). On MRI, it shows low signal intensity on T1-weighted (T1W) images, intermediate to high signal intensity on T2-weighted (T2W) images, and moderate contrast enhancement. Differential diagnosis is mainly with fibrous dysplasia, osteoblastoma, aneurysmal bone cyst, giant cell tumor, and osteosarcoma. On pathology, tumors are composed of fibrocellular tissue and mineralized matrix in variable amounts.


The best treatment option is wide, en bloc tumor resection, which is not always possible in the skull base. Recurrences are not uncommon, particularly after curettage or simple tumor enucleation.
Cartilage-forming tumors
Chondroma , enchondroma , or osteochondroma are relatively common benign cartilaginous neoplasms that may be sporadic or syndromic; the latter is associated with Ollier or Maffuci disease. They are more commonly diagnosed in childhood and early adulthood, with a peak incidence between 10 and 30 years of age. In the skull base, however, these tumors are exceedingly rare and may originate not only from chondrocytic cells in cranial sutures but also from the dura, brain parenchyma, ventricular system, and choroid plexus, resulting from chondroid metaplasia of immature embryonic cells. From the few cases reported in the literature the most common location is the sphenoethmoid region and clivus, but they can occur in any bone originating from endochondral ossification. In the skull base, they show a slight female predilection and are most often diagnosed in the third decade. These slow-growing tumors are clinically silent and come to clinical attention only when they are large enough to produce neurologic symptoms, often related to cranial nerves’ dysfunction or increased intracranial pressure. Biopsy is often misleading, as cytologic features suggest a more aggressive lesion (hypercellularity, anisocariosis, and nuclear hyperchromasia), and therefore, histologic analysis of the full specimen is mandatory to reach the final diagnosis. On imaging, they are characterized by a narrow transition zone, well-defined scalloped margins, cortical expansion, presence of chondroid-type calcifications and absence of periosteal reaction, bone marrow or soft tissue edema, or soft tissue mass. Two different tumor types are recognized: type 1 is more homogeneous, isodense, or of mixed density with minimal to moderate enhancement and type 2 is characterized by the presence of a hypodense, cystic central core. On MRI, lesions tend to be lobular and show intermediate to low signal intensity on T1W images and high to mixed signal intensity on T2W images depending on the variable amount of chondroid calcification. Occasionally, they may rupture the cortex and show an exophytic growth into the surrounding structures. Contrast enhancement is mainly peripheral and along the tumoral septa. In the skull base the main differential diagnoses are with chondrosarcoma and other chondroid tumors, chordoma and meningioma. On pathology, mature hyaline cartilage, variable amounts of calcifications, and a thick fibrous capsule are typical for this lesion. Immunostaining is positive for protein S100 and negative for cytokeratins and EMA (epithelial membrane antigen).
In the context of multiple enchondromatosis syndromes the diagnosis is usually straightforward and the risk of malignant degeneration into a chondrosarcoma is higher, particularly in Maffuci disease (25%–30% in Ollier disease and over 90% in Maffuci disease). In other body locations, the presence of pain outside the context of a pathologic fracture is suspicious for malignant degeneration, but this rule is difficult to apply to the skull base.
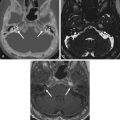
Chondroblastomas are epiphyseal tumors of long bones, thought to arise from epiphyseal growth plates, subarticular in location and quite rare in the head and neck region. In the skull base, the most common location is the temporal bone squamosa, accounting for its endochondral ossification. From there, tumors can grow into the middle cranial fossa, temporomandibular joint (TMJ), and middle ear cavity. Clinical symptoms are often related to hearing and balance dysfunction, such as hearing loss, tinnitus, and otalgia, as well as swelling of the temporal fossa. Seizures can also occur as a consequence of temporal lobe compression and cortical irritation. On imaging, these benign, locally aggressive tumors manifest as expansive lytic lesions, sharply delineated from the adjacent structures by a thin sclerotic rim, with a fuzzy and rarefied appearance, containing variable areas of calcification. On MRI, the tumor matrix tends to be hyperintense on T2W images, reflecting the presence of hyaline cartilage, with variable areas of low T1W and T2W signal intensity reflecting chondroid-like calcifications ( Fig. 16.6 ). Associated aneurysmal bone cysts are present in one-third of cases, shown as expansive lytic areas with fluid-fluid levels. Adjacent synovitis with joint effusion in the adjacent TMJ, periosteal reaction, and bone marrow edema may also be seen.

On imaging, the main differential diagnosis is with other expansive lytic lesions with a bubbly appearance, such as giant cell tumor, aneurysmal bone cyst, and hypervascular metastases, and with other lesions containing chondroid matrix, such as chondromyxoid fibroma and chondrosarcoma. On pathology, chondroblastomas are composed of polyhedral chondroblasts, within a reticulin network showing pericellular calcifications in a typical “chicken wire” pattern. Scattered osteoclast-like giant cells surround foci of chondroid matrix. Immunohistochemistry is mandatory for the differential diagnosis: chondroblastomas show immunopositivity for protein S100, whereas the main lesions to be considered in the differential, both on imaging and pathologic grounds, do not. Although this is mainly a benign tumor, rare cases of malignant chondroblastomas have been reported. In the skull base, radical excision with eventual bone grafting rather than curettage is the treatment of choice associated with a decreased incidence of tumor recurrence (43% in the former vs. 27% in the latter). In cases of incomplete resection, radiation therapy should be offered to these patients.
Chondromyxoid fibroma is a rare, slow-growing, predominantly metaphyseal tumor, found in young adults in the second and third decades of life, being exceedingly rare in the craniofacial region, with only 67 cases described in the English literature. Although this tumor is benign, it is often mistaken for a more aggressive lesion because of its infiltrative/destructive appearance, and it is prone to local recurrence when incompletely resected, a common occurrence in skull base lesions. On CT, these tumors manifest as an osteolytic, lobulated, expansive lesion often surrounded by a sclerotic margin, occasionally with discrete calcification of the tumor matrix (seen in 25%–75% of cases), resembling a nonossifying fibroma. On MR, they show low signal intensity on T1W and heterogenous contrast enhancement after gadolinium administration. When fibrous tissue predominates, lesions show low to intermediate signal intensity on T2W images, whereas when a chondroid or myxoid matrix is dominant, lesions are T2 hyperintense ( Fig. 16.7 ). The main differential diagnosis is with other myxoid tumors: chordoma, chondroid chordoma, and chondrosarcoma, which are much more commonly seen in the skull base. Pathologically, chondromyxoid fibromas are characterized by the presence of lobules of spindle-shaped or stellate cells in a background of abundant myxoid or chondroid intercellular material, containing multinucleated giant cells. Complete surgical resection is the treatment of choice, although often impossible in skull base tumors lying close to vital neurovascular structures. The benefit of postoperative radiation treatment for irresectable, residual, and recurrent disease is controversial.

Tumors without osteoid or chondroid matrix
Intraosseous hemangiomas are benign vascular lesions originating from the bone marrow. The most common locations are the vertebrae and calvaria, particularly the frontal and parietal bones, with skull base hemangiomas being exceedingly rare. In the craniofacial region, the maxilla, zygoma, vomer, and mandible are most commonly affected, and those lesions involving the orbital walls are called primary intraosseous orbital hemangiomas. Hemangiomas have also been reported in the temporal bone, most often in the region of the geniculate fossa, and the main differential diagnosis is with facial nerve hemangiomas, which originate from the perineural vascular plexus rather than the petrous bone proper. In the skull base, only 33 lesions have been reported so far, affecting the occipital condyle, sphenoorbital region, and greater sphenoid wing.
Hemangiomas tend to be confined to the bone marrow cavity, with atypical or more aggressive hemangiomas having an associated soft tissue component. These are slow-growing, commonly asymptomatic lesions found incidentally on unrelated imaging studies, but occasionally, they can cause headaches and craniofacial symptoms secondary to compression of neighboring structures. On CT imaging, they present as lytic lesions surrounded by a thin sclerotic rim. When these lesions contain coarse bony trabeculae, arranged from the center to the periphery, in a typical honeycomb/soap-bubble/sunburst pattern, they are called ossifying hemangiomas. Hemangiomas can become large enough to expand and remodel cortical bone and lead to areas of dehiscence with associated soft tissue components. However, as opposed to osteogenic osteosarcomas, the periosteal lining tends to remain intact and the soft tissue component does not invade adjacent structures. On MR, lesions are mottled in appearance, with variable signal intensity on T1W, often hyperintense on T2W and showing delayed, centripetal contrast-enhancement after gadolinium administration. Areas of high signal intensity on T1W images reflect the presence of fatty marrow or fresh thrombus.
In the skull base, they are often misdiagnosed for other more common lesions, and therefore the diagnosis is frequently made during exploratory surgery.
On pathology, depending on the type of vascular network, they are classified into capillary, cavernous, or venous hemangiomas and are composed of a tangle of endothelially lined vessels together with variable amounts of thrombus, fat, fibrous tissue, and smooth muscle.
Treatment is required only for symptomatic lesions leading to mass effect or bleeding and includes embolization, sclerotherapy, surgical excision, and/or radiation therapy.
Aneurysmal bone cyst is a lesion of uncertain etiology, composed of numerous blood-filled spaces without endothelial lining, separated by bone trabeculae, connective tissue, osteoid, and osteoclasts. These lesions can be primary or secondary to other bone lesions such as osteoblastoma, chondroblastoma, fibrous dysplasia, giant cell tumor, and osteosarcoma and, therefore, they are often considered reactive lesions ( Fig. 16.3 ). Primary lesions are most often seen in young adults, around 80% before age 20 years. These lesions are rare in the craniofacial region; the most common location is the basisphenoid and paranasal sinuses. On imaging, the hallmark of this lesion is the presence of fluid-fluid levels, best appreciated on MRI owing to the sedimentation effect of blood degradation products, showing variable signal intensity. Lesions display a hemosiderin peripheral rim, hyperintense on both T1W and T2W images, with susceptibility artifact on T2∗ or susceptibility weighted imaging, and internal septa, which may enhance after gadolinium administration. On CT, they present as a well-delineated, expansive, soap-bubbly multicystic lesion, with thin sclerotic margins without periosteal reaction unless complicated by fracture. It should be noted, however, that many other lesions may show fluid-fluid levels, such as giant cell tumors, osteoblastoma, chondroblastoma, and osteosarcoma (particularly the telangiectatic subtype), just to mention the most common. Treatment is by curettage or complete surgical resection when possible. Preoperative embolization may help reduce tumor size and decrease surgical bleeding.
Giant cell tumors or osteoclastomas originate from nonosteogenic stromal cells within the bone marrow cavity. They are part of the spectrum of giant cell lesions that also include giant cell granulomas and brown tumors and result from an overproliferation of osteoclasts. This is a tumor of adulthood more commonly seen between 20 and 50 years of age, after growth plate closure. In long bones, they are typically epiphyseal, are subarticular in location, and present clinically with pain and local swelling. The most common locations in the skull base are the sphenoid and temporal bones. Associated symptoms result from direct involvement or compression of adjacent structures and therefore they depend on on the exact location of the lesion and its size. Hearing loss and cranial nerve and TMJ dysfunction are among the most frequent symptoms. Pathologically, giant cell tumors are characterized by the presence of multinucleated giant cells, spindle-shaped mononuclear cells, cysts, and multiple vessels that are prone to hemorrhage, accounting for the presence of hemosiderin-laden macrophages responsible for the increased density of these lesions on plain CT scans.
On imaging studies, giant cell tumors, appear as sharply marginated, scalloped, lytic lesions, expanding and remodeling cortical bone, sometimes leading to areas of bony dehiscence, without sclerotic rim and mineralized or calcified matrix. MR features are variable depending on the degree of hemorrhage and presence of aneurysmal bone cyst component, which can be clearly differentiated on contrast-enhanced T1W images: solid tumor components enhance strongly, whereas the cystic components show only faint peripheral enhancement after gadolinium administration. Spontaneously T1W hyperintensities may be seen and reflect the presence of subacute bleeding. T2W images can also be used to differentiate hyperintense cystic components, often with fluid-fluid levels from intermediate to hypointense solid tumor components. A hypointense rim of hemosiderin is the rule blending with the cortical margin of the lesion ( Fig. 16.8 ).