To keep up with advances in central nervous system (CNS) tumor diagnosis and discovery of new entities, the classification of these tumors requires periodic review and revision. Since the initial 1979 publication from the World Health Organization (WHO) of Histological Typing of Tumours of the Central Nervous System, 3 further editions have been published, cataloging the advances in CNS tumor classification and diagnosis over the past 3 decades. In this article, we discuss select new additions to the current classification, including new diagnostic tools, differential diagnoses, and management implications.
To keep up with advances in central nervous system (CNS) tumor diagnosis and discovery of new entities, the classification of these tumors requires periodic review and revision. Since the initial 1979 publication from the World Health Organization (WHO) of Histological Typing of Tumours of the Central Nervous System, 3 further editions have been published, cataloging the advances in CNS tumor classification and diagnosis over the past 3 decades. The second edition, published in 1993, incorporated advances in tumor classification resulting from the use of immunohistochemical techniques in diagnosis. Subsequently, the third edition, published in 2000, elaborated on the use of genetic techniques as an aid to tumor diagnosis, as well as describing associated clinical and radiologic findings and prognostic factors. The current 2007 edition includes several new entities, variants, and patterns of differentiation, and expands on the use of molecular techniques in diagnosis and prognostication. In this article, we discuss select new additions to the current classification, including new diagnostic tools, differential diagnoses, and management implications.
Angiocentric glioma, WHO grade 1
Angiocentric glioma is a low-grade indolent cortically based tumor of the cerebrum usually presenting in children with a history of seizures. The tumor was first recognized in as a new entity in 2005. The name originates from the presence of perivascular tumor cells, which are mostly bipolar, and are arranged in a circumferential, longitudinal, or perpendicular orientation to blood vessels.
A total of 26 cases had been described at the time of publication of the 2007 WHO Classification of Tumours of the Central Nervous System , with a mean age of 17 years. However, all but 2 of the 26 presented with seizures in childhood. Five cases, all 13 years of age or younger, have been described since. One additional cortical tumor in a 5-year-old presenting with headaches has been reported, with features described as ependymoma or angiocentric glioma. Most of the patients had a preceding history of either refractory epilepsy or childhood seizures, but one case presented with headache, loss of visual acuity, and no seizure history.
Radiographically, the tumor appears as an ill-defined cortical lesion that is hyperintense on T2-weighted magnetic resonance images, and noncontrast enhancing on T1-weighed images, with superficial extension into the associated subjacent white matter. Diffusion tensor imaging tractography has revealed tumor displacement of fibers. One case has been reported to contain microcalcifications not detected radiographically. The presence of a T2/FLAIR (fluid attenuated inversion recovery) “stalk-like” extension of the cortical tumor extending to the ventricle was described in a case series by Lellouch-Tubiana and colleagues.
Histologically, the tumor is composed of a monomorphic population of cells, most with bipolar or fusiform cell processes, infiltrating the cortex as a single cell population with associated entrapped cortical neurons or as masses of fusiform cells. In less cellular areas, the tumor cells can have a circumferential and longitudinal orientation to blood vessels including capillaries ( Fig. 1 A). In more densely cellular areas, the tumor cells are more radially oriented, forming perivascular pseudorosettes and nodules of compact tumor cells. The tumor is also reported to have subpial spread, with the tumor cells oriented perpendicular to the pial surface. The cells are variably glial fibrillary acidic protein (GFAP) and EMA immunopositive and typically vimentin immunopositive (see Fig. 1 B, C). When positive, EMA usually has a dotlike immunopositive staining pattern similar to ependymomas, but unlike ependymomas, angiocentric glioma tumor cells are CD99 immunonegative. The tumor is variably immunopositive for neuronal markers, such as synaptophysin, NeuN, and chromogranin.
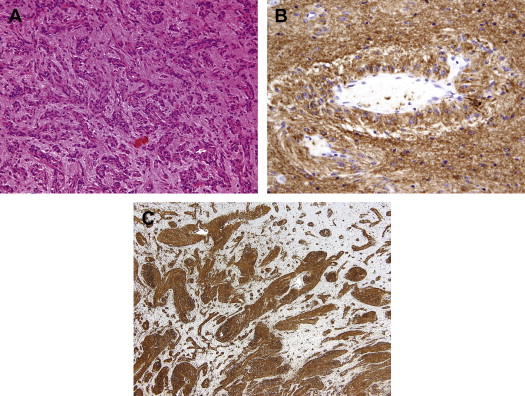
Electron microscopy of some cases has revealed focal microlumina with associated microvilli and “zipper-like” intracellular junctions, suggesting an ependymal phenotype. Preusser and colleagues postulate an origin from radial glia, the basis for this theory being the radial orientation of tumor cells within the cortex, and the immunopositivity of the tumor for GFAP, vimentin, and S100. Chromosomal comparative genomic hybridization analysis of tumor DNA has revealed a loss of chromosomal bands 6q24-q25 and gain of chromosomal band 11p11.2.
In one patient, placement of depth electrodes within the tumor revealed ictal activity centered within the tumor before spread to the surrounding cortex, suggesting the utility of tumor depth electrodes to define tumor extent before surgery. Although typically a low-grade tumor, at least one reported case progressed following subtotal resection, with recurrent seizures and death 62 months after initial resection. In contrast, a second case had small polymorphous areas histologically, but showed no evidence of recurrence after 9 months of follow-up. Each of these cases had an elevated MIB-1(Ki-67) labeling index, 10% and 8% respectively. Following gross total or subtotal resection, most patients have been seizure free without progression.
Atypical choroid plexus papilloma, WHO grade 2
Most intraventricular choroid plexus tumors (CPT) are benign and designated choroid plexus papilloma (CPP) with a current WHO classification of Grade 1. CPPs have a low rate of recurrence and do not require adjuvant therapy following gross total resection. Much less common are choroid plexus carcinomas (CPC), which exhibit clear features of malignancy. However, brain invasion is not the sole criteria for malignancy. Diagnosis of CPC requires the presence of 4 of the 5 following atypical histologic features: high mitotic activity, increased cellularity, nuclear pleomorphism, sheeting of tumor cells, and necrosis. CPCs are classified as WHO Grade 3 tumors. A new intermediate category is described in the current WHO classification: atypical choroid plexus papilloma (APP), WHO Grade 2, which has an uncertain clinical prognosis. Childhood CPTs most commonly occur in the lateral ventricles, whereas adult CPTs most commonly occur in the posterior fossa. CPTs account for 0.4% to 1.0% of adult intracranial tumors, but 4.0% of pediatric intracranial tumors. APPs have been found to occur at a significantly younger age (median age of 0.7 years) than either CPPs or CPCs (both with a median age of 2.3 years). The main feature distinguishing APP from CPP is an elevated mitotic activity defined as equal to or greater than 2 per 10 high-powered fields. Mitotic count is the only histologic feature independently associated with tumor recurrence, while achievement of gross total resection is the main clinical feature impacting the likelihood of tumor recurrence. Malignant progression in choroid plexus papillomas is rare, but there is at least one report of an APP occurring as a recurrence after a 15-year dormant period following resection of a CPP. A recent study of 106 patients with choroid plexus tumors revealed a 5-year event-free survival of 92%, 83%, and 28% for CPP, APP, and CPC respectively. APPs have a risk of metastasizing almost equal to CPCs, whereas CPPs metastasize at a rate of 5%. The clinical symptoms of choroid plexus tumors include headache, nausea, vomiting, altered visual acuity, and papilledema, commonly attributable to the presence of hydrocephalus.
Generally, magnetic resonance imaging (MRI) is considered the best choice for diagnosis of CPTs, the tumors appearing as homogeneous or heterogeneous, hyperdense or hyperintense masses that are contrast enhancing ( Fig. 2 A, B).
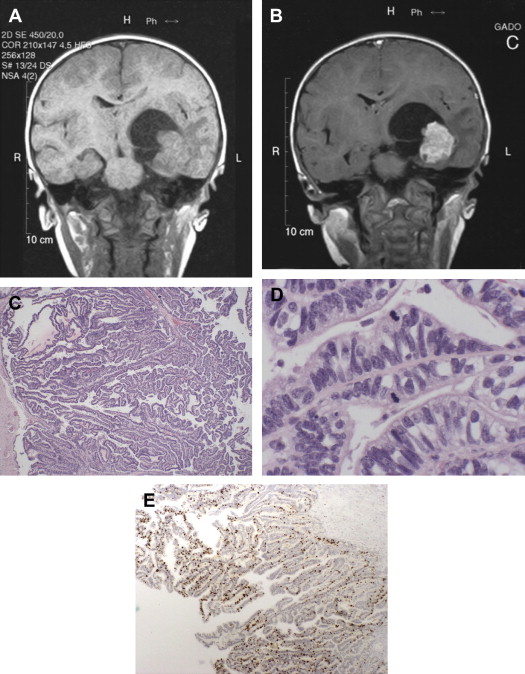
Histologically, APPs are composed of fibrovascular papillary structures having a simple layer of cuboidal or columnar epithelial cells with slight nuclear atypia, sometimes in a pseudo-columnar architecture. The thin fibrovascular papillary fronds tend to maintain their normal papillary architecture, but present with at least one atypical feature, most commonly elevated mitotic activity (see Fig. 2 C, D). Immunohistologically, the tumor cells are typically immunopositive for cytokeratin, GFAP, S100, and TTR, but immunonegative for carcinoembryonic antigen (CEA). Immunohistochemical staining for p53 and MIB-1 may be helpful in determining the tumor grade. Both CPPs and APPs are typically immunonegative for p53 (0% and 9.5% respectively), whereas CPCs are typically immunopositive (47.1%). Immunohistochemical stains for MIB-1 reveal an increasing labeling index with increasing grade (1.3, 9.1, and 20.3 for CPPs, APPs, and CPCs respectively) (see Fig. 2 E).
Genetic analysis of APPs has revealed variable cytogenetic and genomic abnormalities, including normal chromosomal karyotype. Most studies have demonstrated normal or hyperdiploid karyotypes for CPCs.
The histologic grade of CPTs is important because CPPs tend not to recur or need treatment with adjuvant therapy, although long-term follow-up is still recommended. In contrast, although APPs have a higher rate of incomplete resection and metastasis than CPPs, they appear to respond favorably to chemotherapy with complete, partial, or stable remission in most cases.
Atypical choroid plexus papilloma, WHO grade 2
Most intraventricular choroid plexus tumors (CPT) are benign and designated choroid plexus papilloma (CPP) with a current WHO classification of Grade 1. CPPs have a low rate of recurrence and do not require adjuvant therapy following gross total resection. Much less common are choroid plexus carcinomas (CPC), which exhibit clear features of malignancy. However, brain invasion is not the sole criteria for malignancy. Diagnosis of CPC requires the presence of 4 of the 5 following atypical histologic features: high mitotic activity, increased cellularity, nuclear pleomorphism, sheeting of tumor cells, and necrosis. CPCs are classified as WHO Grade 3 tumors. A new intermediate category is described in the current WHO classification: atypical choroid plexus papilloma (APP), WHO Grade 2, which has an uncertain clinical prognosis. Childhood CPTs most commonly occur in the lateral ventricles, whereas adult CPTs most commonly occur in the posterior fossa. CPTs account for 0.4% to 1.0% of adult intracranial tumors, but 4.0% of pediatric intracranial tumors. APPs have been found to occur at a significantly younger age (median age of 0.7 years) than either CPPs or CPCs (both with a median age of 2.3 years). The main feature distinguishing APP from CPP is an elevated mitotic activity defined as equal to or greater than 2 per 10 high-powered fields. Mitotic count is the only histologic feature independently associated with tumor recurrence, while achievement of gross total resection is the main clinical feature impacting the likelihood of tumor recurrence. Malignant progression in choroid plexus papillomas is rare, but there is at least one report of an APP occurring as a recurrence after a 15-year dormant period following resection of a CPP. A recent study of 106 patients with choroid plexus tumors revealed a 5-year event-free survival of 92%, 83%, and 28% for CPP, APP, and CPC respectively. APPs have a risk of metastasizing almost equal to CPCs, whereas CPPs metastasize at a rate of 5%. The clinical symptoms of choroid plexus tumors include headache, nausea, vomiting, altered visual acuity, and papilledema, commonly attributable to the presence of hydrocephalus.
Generally, magnetic resonance imaging (MRI) is considered the best choice for diagnosis of CPTs, the tumors appearing as homogeneous or heterogeneous, hyperdense or hyperintense masses that are contrast enhancing ( Fig. 2 A, B).
Histologically, APPs are composed of fibrovascular papillary structures having a simple layer of cuboidal or columnar epithelial cells with slight nuclear atypia, sometimes in a pseudo-columnar architecture. The thin fibrovascular papillary fronds tend to maintain their normal papillary architecture, but present with at least one atypical feature, most commonly elevated mitotic activity (see Fig. 2 C, D). Immunohistologically, the tumor cells are typically immunopositive for cytokeratin, GFAP, S100, and TTR, but immunonegative for carcinoembryonic antigen (CEA). Immunohistochemical staining for p53 and MIB-1 may be helpful in determining the tumor grade. Both CPPs and APPs are typically immunonegative for p53 (0% and 9.5% respectively), whereas CPCs are typically immunopositive (47.1%). Immunohistochemical stains for MIB-1 reveal an increasing labeling index with increasing grade (1.3, 9.1, and 20.3 for CPPs, APPs, and CPCs respectively) (see Fig. 2 E).
Genetic analysis of APPs has revealed variable cytogenetic and genomic abnormalities, including normal chromosomal karyotype. Most studies have demonstrated normal or hyperdiploid karyotypes for CPCs.
The histologic grade of CPTs is important because CPPs tend not to recur or need treatment with adjuvant therapy, although long-term follow-up is still recommended. In contrast, although APPs have a higher rate of incomplete resection and metastasis than CPPs, they appear to respond favorably to chemotherapy with complete, partial, or stable remission in most cases.
Papillary glioneuronal tumor, WHO grade 1
The entity “papillary glioneuronal tumor” (PGNT) was first defined by Komori and colleagues, who described a mixed glial and neuronal tumor with indolent behavior and good prognosis, which is characterized as WHO grade 1. The tumor has been reported in patients from 7 to 75 years of age, but most tumors present in young adults. PGNTs typically present as cerebral lesions with both cystic and solid areas and can be large, reportedly up to 9 cm. The tumors have a greater than 7-year progression-free survival following gross total resection. PGNTs typically do not have atypical features such as high mitotic activity, necrosis, and vascular hyperplasia/endothelial hyperplasia. However, PGNTs with elevated proliferation rates and aggressive behavior have been reported. Two recently reported cases of aggressive PGNTs had an initial MIB-1 labeling index of 1% and 4% respectively, and rapidly recurred despite gross total resection. Another reported case had an elevated labeling index of 10%, and had mini-gemistocytic cells; this tumor rapidly progressed despite partial resection and adjuvant radio-chemotherapy. However, a case reported by Vaquero and Coca in 2006 had an elevated MIB-1 labeling index of 15% and was symptom-free 5 years after radical surgery and radiotherapy, indicating that high proliferation rate alone does not always predict aggressive behavior. PGNTs are associated with focal hemorrhage and there is at least one reported case of progressive superficial siderosis attributable to chronic subarachnoid hemorrhage.
Clinically, PGNTs commonly present with mild neurologic symptoms such as headache and vision changes, but the tumors can also present with nausea and vomiting, seizures, or hemiparesis. Asymptomatic cases discovered incidentally have also been reported.
Radiographically, the tumors are most commonly found to have large uniloculated or multiloculated cysts with an associated mural nodule. On MRI, the tumors are typically hypointense on T1, hyperintense on T2, and usually show enhancement of the mural nodule and sometimes enhancement of the cyst wall ( Fig. 3 A–C). Typically, PGNTs have little associated edema or mass effect.
Histologically, the tumors are composed of compact areas of pseudopapillary-appearing thick hyalinized blood vessels lined by 1 or 2 cell layers of small astrocytic tumor cells with round uniform nuclei (see Fig. 3 D). These astrocytic cells are immunopositive for GFAP and S100. Surrounding the astrocytic cells are oligodendrocytic cells with round nuclei and scant to indistinct cytoplasm that are immunopositive for Olig2, a nonspecific marker for oligodendrocyte differentiation. The cells within the spaces between the pseudopapillary vessels are composed of variably small and medium-sized neuronal tumor cells and, when present, large ganglion tumor cells. These small, medium, and large neuronal cells are each immunopositive for neuronal markers such as synaptophysin, NeuN, neurofilament (NF), and neuron specific enolase (NSE). Most PGNTs have no necrosis, few if any areas of vascular proliferation, a low MIB-1 proliferation index (<1%), and most of the cases have been p53 immunonegative. Rosenthal fibers, eosinophilic granular bodies, hemosiderin, and calcifications are commonly present at the periphery of the tumors.
Electron microscopy has confirmed the presence of microfilaments within glial processes, and microtubules within immature neurons. One report of chromosomal analysis of a PGNT identified genetic alteration of chromosome 7 alone, with breakpoints at 7p22. No deletions of chromosomes 1p or 19q have been reported.
Although PGNTs tend to have good prognosis with no need for adjuvant therapy following gross total resection, reports of atypical and aggressive PGNTs suggest the need for ongoing careful reporting of atypical features for future therapeutic implications.
Rosette-forming glioneuronal tumor of the fourth ventricle, WHO grade 1
This tumor is a mixed glial-neuronal neoplasm affecting predominantly young adults, and centered on the region of the fourth ventricle. The tumor was originally described as a distinct entity by Komori and colleagues in 2002 in a series of 11 posterior fossa tumors. Before this, a tumor with the same histologic appearance was described as dysembryoplastic neuroepithelial tumor of the cerebellum by Kuchelmeister and colleagues in 1995. At the time of the current WHO guide going to press, 17 cases had been reported. Since then, a further 12 cases have been described. One of these cases had the characteristic histomorphology, but was located in the optic chiasm and left optic nerve of a patient with neurofibromatosis type 1.
Rosette-forming glioneuronal tumor of the fourth ventricle (RGNT) occurs over an age range of 12 to 59 years, with a possible slight female predilection. Patients typically present with headaches and ataxia as a result of obstructive hydrocephalus, and may occasionally experience neck pain. RGNT has a midline location, involving the fourth ventricle and/or aqueduct. At the periphery, the tumor may also involve the cerebellar vermis, adjacent brain stem, thalamus, or pineal gland. The tumor may be multicentric.
Upon MRI, the tumor is relatively circumscribed and is mostly solid with heterogeneous areas, the solid portions of the tumor being iso- to hypointense on T1-weighted imaging and hyperintense on T2 imaging. It may be partially cystic or multiloculated, and dense calcifications have been seen in some examples. Multicentric, flaccid curvilinear or ring enhancement appears to be characteristic for RGNT. In contrast, the ring enhancement that can be seen in pilocytic astrocytomas is usually solitary. Radiologically, distinction of RGNT from medulloblastoma (which can also have a similar location) can be achieved because medulloblastomas have high density on computed tomography (CT) and diffusion restriction on MRI, reflecting the high cellularity of this tumor.
Histologically, the biphasic nature of RGNT is reflected in its distinct glial and neurocytic components ( Fig. 4 A). The tumor has an overall low cellularity. The glial component closely resembles pilocytic astrocytoma, composed of elongated “piloid” glial cells within a variably compact to loose fibrillary background, sometimes with microcystic areas (see Fig. 4 B). Also like pilocystic astrocytoma, there may admixed Rosenthal fibers, eosinophilic granular bodies (EGBs), microcalcifications, and hemosiderin deposits. Similarly, thick-walled hyalinized vessels and glomeruloid vascular structures may be present. Therefore, recognition of the accompanying neurocytic component is important to avoid confusion with pilocytic astrocytoma. The “neurocytic” portions of the tumor contain cells with round nuclei and finely speckled chromatin, arranged in perivascular pseudorosettes and small neurocytic rosettes surrounding a central core of neuropil (see Fig. 4 C). There may be an interspersed mucinous matrix with microcysts. Occasional ganglion cells may be seen. As may be expected, the neuropil matrix is immunopositive for synaptophysin, as are the neurocytic cells (see Fig. 4 D). There is similar labeling of these cells for MAP-2 and NSE. The glial component is labeled by immunostains for GFAP and S100 protein. Depending on plane of section, the neurocytic component can histologically resemble dysembryoplastic neuroepithelial tumor (DNT), with perivascular columns of tumor cells suspended within a mucoid matrix.
Related posts:
 Molecular Tools: Biology, Prognosis, and Therapeutic Triage
Molecular Tools: Biology, Prognosis, and Therapeutic Triage
 Advanced Imaging in Brain Tumor Surgery
Advanced Imaging in Brain Tumor Surgery
 Imaging of Brain Tumors: Perfusion/Permeability
Imaging of Brain Tumors: Perfusion/Permeability
 Novel Medical Therapeutics in Glioblastomas, Including Targeted Molecular Therapies, Current and Future Clinical Trials
Novel Medical Therapeutics in Glioblastomas, Including Targeted Molecular Therapies, Current and Future Clinical Trials
 Clinical Trials in Brain Tumor Surgery
Clinical Trials in Brain Tumor Surgery
 Imaging of Brain Tumors: Functional Magnetic Resonance Imaging and Diffusion Tensor Imaging
Imaging of Brain Tumors: Functional Magnetic Resonance Imaging and Diffusion Tensor Imaging
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


