Systemic and Lung Diseases
In this chapter, examples are used to illustrate how certain systemic and lung diseases, not already covered in previous chapters, can manifest and affect the appearance of the pulmonary vasculature. The diseases explored here include sarcoidosis, hepatopulmonary syndrome, sickle cell disease, bronchopulmonary dysplasia, idiopathic pulmonary hemosiderosis, emphysema, pulmonary Langerhans cell histiocytosis, usual interstitial pneumonitis, lymphangiomatosis and the angiomatous diseases, which include capillary hemangiomatosis and pulmonary capillary veno-occlusive disease.
 Sarcoidosis
Sarcoidosis
 Sarcoidosis is a systemic noncaseating granulomatous disease of unknown etiology that affects the mediastinum, lungs, and other organs to varying degrees.
Sarcoidosis is a systemic noncaseating granulomatous disease of unknown etiology that affects the mediastinum, lungs, and other organs to varying degrees.
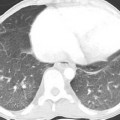
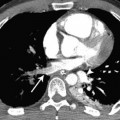
 In its most severe form, sarcoidosis can manifest as fibrosing mediastinitis with obliteration of the pulmonary arteries (Fig. 11.1) and veins (Fig. 11.2).
In its most severe form, sarcoidosis can manifest as fibrosing mediastinitis with obliteration of the pulmonary arteries (Fig. 11.1) and veins (Fig. 11.2).
 Pulmonary hypertension occurs in patients with end-stage lung disease due to sarcoidosis; several mechanisms may contribute, including fibrotic destruction of the capillary bed and resultant chronic hypoxemia, extrinsic compression of the major pulmonary arteries by enlarged lymph nodes, and secondary pulmonary veno-occlusive disease.
Pulmonary hypertension occurs in patients with end-stage lung disease due to sarcoidosis; several mechanisms may contribute, including fibrotic destruction of the capillary bed and resultant chronic hypoxemia, extrinsic compression of the major pulmonary arteries by enlarged lymph nodes, and secondary pulmonary veno-occlusive disease.
 Hepatopulmonary Syndrome
Hepatopulmonary Syndrome
 Hepatopulmonary syndrome is defined as the triad of hepatic dysfunction, intrapulmonary vascular dilatation, and hypoxemia.
Hepatopulmonary syndrome is defined as the triad of hepatic dysfunction, intrapulmonary vascular dilatation, and hypoxemia.
 Clinically, it manifests as progressive dyspnea in patients with cirrhosis.
Clinically, it manifests as progressive dyspnea in patients with cirrhosis.
 It occurs in 50% of patients with chronic liver disease of any cause.
It occurs in 50% of patients with chronic liver disease of any cause.
 It is not to be confused with portopulmonary hypertension, which is pulmonary hypertension due to increased vascular resistance in patients with portal hypertension.
It is not to be confused with portopulmonary hypertension, which is pulmonary hypertension due to increased vascular resistance in patients with portal hypertension.
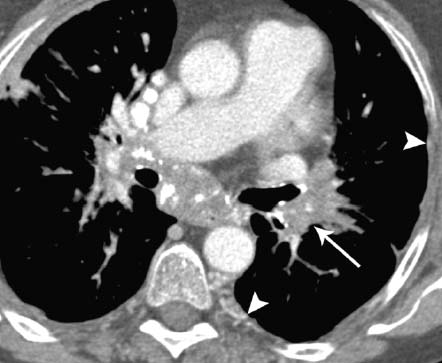
Fig. 11.1 Fibrosing mediastinitis due to sarcoidosis in a 72-year-old man. Axial CT demonstrates extensive mediastinal and hilar lymphadenopathy. There is obliteration of the left lower lobe pulmonary artery by the fibrotic mass (arrow) and intercostal artery collaterals (arrowheads).
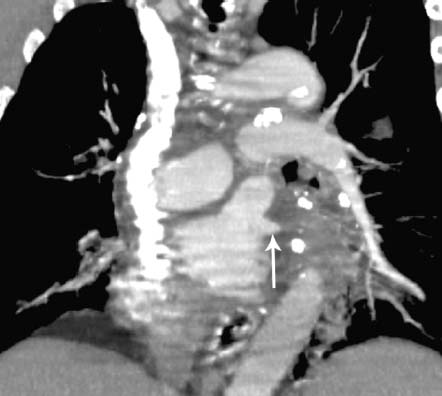
Fig. 11.2 Fibrosing mediastinitis due to sarcoidosis in a 61-year-old man. Coronal reconstruction CT demonstrates obliteration of the left inferior pulmonary vein (arrow) secondary to sarcoidosis.
 Normal pulmonary capillaries are between 8 and 15 [H9262]m in diameter; in hepatopulmonary syndrome, they are 15 to 100 [H9262]m in diameter.
Normal pulmonary capillaries are between 8 and 15 [H9262]m in diameter; in hepatopulmonary syndrome, they are 15 to 100 [H9262]m in diameter.
 The hypoxemia is due to the presence of numerous tiny intrapulmonary shunts.
The hypoxemia is due to the presence of numerous tiny intrapulmonary shunts.
 Radiographic findings include bilateral basilar nodular or reticular opacities on chest radiograph, heterogeneous uptake on perfusion lung scan, and peripheral arteriolar dilatation on computed tomography (CT) or magnetic resonance imaging (MRI; Fig. 11.3).
Radiographic findings include bilateral basilar nodular or reticular opacities on chest radiograph, heterogeneous uptake on perfusion lung scan, and peripheral arteriolar dilatation on computed tomography (CT) or magnetic resonance imaging (MRI; Fig. 11.3).
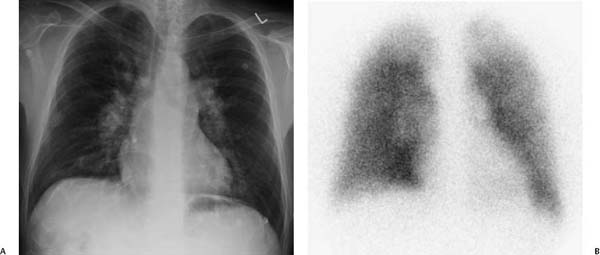
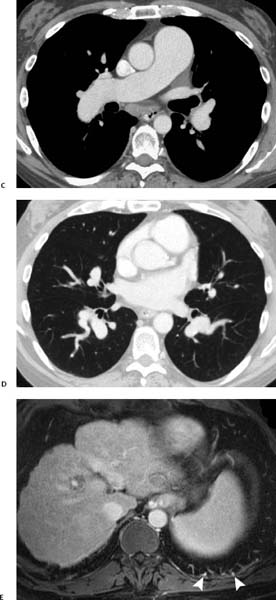
Fig. 11.3 (A–E) Hepatopulmonary syndrome in a 47-year-old man. (A) Chest radiograph demonstrates a heart of normal size with dilated central and peripheral pulmonary arteries. (B) Technetium Tc 99m macroaggregate (99mTc-macroaggregate) albumin perfusion scan demonstrates heterogeneous pulmonary perfusion with no segmental defects. The ventilation scan result was normal. Hepatopulmonary syndrome in a 47-year-old man. (C) CT demonstrates dilated main, right, and left pulmonary arteries. (D) CT demonstrates dilated segmental arteries (the diameter of the artery is much larger than the diameter of the accompanying bronchus); also, there are dilated and tortuous subsegmental arteries. (E) MRI shows dilated subpleural pulmonary vessels (arrowheads). In addition, a small nodular liver and esophageal varices are seen.
 Sickle Cell Disease
Sickle Cell Disease
 Sickle cell disease is a systemic disease that can result in occlusion of the small pulmonary vessels.
Sickle cell disease is a systemic disease that can result in occlusion of the small pulmonary vessels.
 In the acute chest syndrome, capillary obstruction by sickle cells is accompanied by in situ thrombosis.
In the acute chest syndrome, capillary obstruction by sickle cells is accompanied by in situ thrombosis.
 Imaging can demonstrate ground-glass opacification or extensive consolidation due to hemorrhagic edema, which is caused by ischemia, infarction, or infection and is responsible for precipitation of the acute chest syndrome (Fig. 11.4).
Imaging can demonstrate ground-glass opacification or extensive consolidation due to hemorrhagic edema, which is caused by ischemia, infarction, or infection and is responsible for precipitation of the acute chest syndrome (Fig. 11.4).
 CT can show mosaic attenuation, thought to be related to secondary pulmonary lobules with hypoperfusion adjacent to normal or hyperperfused lobules (Fig. 11.4).
CT can show mosaic attenuation, thought to be related to secondary pulmonary lobules with hypoperfusion adjacent to normal or hyperperfused lobules (Fig. 11.4).
 Chronic changes are seen in individuals experiencing repeated acute episodes, with parenchymal bands due to fibrosis of infarctions (Fig. 11.4).
Chronic changes are seen in individuals experiencing repeated acute episodes, with parenchymal bands due to fibrosis of infarctions (Fig. 11.4).
 Eventually, in patients with persistent occlusion of the microvascular bed, cor pulmonale develops, manifested as dilatation of the pulmonary arteries and right ventricular hypertrophy.
Eventually, in patients with persistent occlusion of the microvascular bed, cor pulmonale develops, manifested as dilatation of the pulmonary arteries and right ventricular hypertrophy.
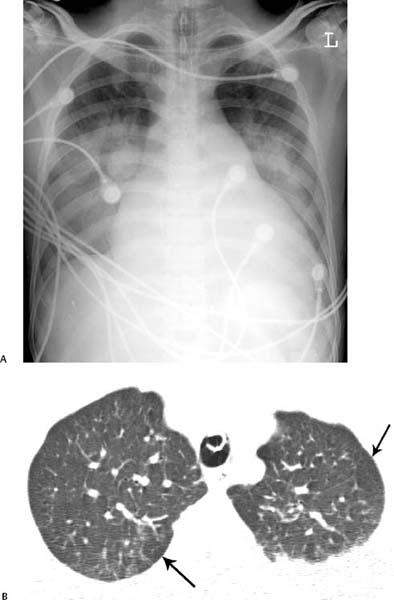
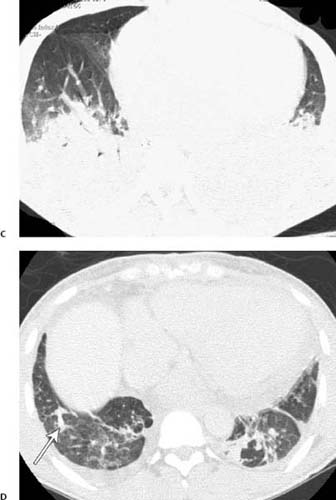
Fig. 11.4 (A–D) Sickle cell crisis in a 27-year-old man. (A) Chest radiograph demonstrates that the patient is intubated. Even allowing for the projection, the heart appears enlarged and the main pulmonary artery prominent. There is bilateral consolidation, which can represent pulmonary infarction or precipitating pneumonia. (B) CT performed at the same time as chest radiography demonstrates bilateral upper lobe mosaic attenuation. The more lucent regions represent poorly perfused lobules (arrows). Sickle cell crisis in a 27-year-old man. (C) More inferiorly, CT demonstrates bilateral lower lobe consolidation. (D) CT performed at a later date demonstrates bilateral linear lung scars, manifested as bands (arrow), as well as a persistent mosaic pattern of lung attenuation.
 Bronchopulmonary Dysplasia
Bronchopulmonary Dysplasia
 In premature neonates, the high pressures of oxygen delivery can result in necrotizing vasculitis, bronchiolitis, and alveolar septal injury.
In premature neonates, the high pressures of oxygen delivery can result in necrotizing vasculitis, bronchiolitis, and alveolar septal injury.
 CT features in older survivors include extensive areas of decreased lung attenuation, within which the size and number of vessels and bronchi are reduced as a consequence of chronic changes in the lung parenchyma, vessels, and airways (Fig. 11.5).
CT features in older survivors include extensive areas of decreased lung attenuation, within which the size and number of vessels and bronchi are reduced as a consequence of chronic changes in the lung parenchyma, vessels, and airways (Fig. 11.5).
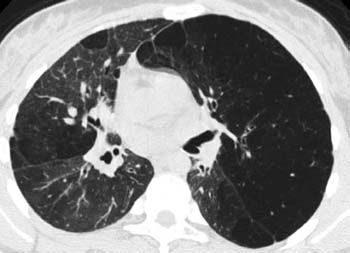
Fig. 11.5 Bronchopulmonary dysplasia in a 13-year-old girl. High-resolution CT demonstrates a mosaic pattern of lung attenuation. The more lucent regions of lung have small vessels, consistent with chronic vessel damage or shunting of blood to the more attenuating and functional regions of lung. There is also shift of the mediastinum from left to right secondary to a large left lung volume resulting from air trapping. The bronchi and bronchioles in the lucent regions of lung are also noted to be small in diameter.
 Idiopathic Pulmonary Hemosiderosis
Idiopathic Pulmonary Hemosiderosis
 Idiopathic pulmonary hemosiderosis is a rare cause of diffuse alveolar hemorrhage and, by definition, of unknown etiology.
Idiopathic pulmonary hemosiderosis is a rare cause of diffuse alveolar hemorrhage and, by definition, of unknown etiology.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


